PF-04691502 is a novel, potent, ATP-competitive and selective dual inhibitor of PI3K (phosphatidylinositol 3 kinase) and mTOR (mammalian target of rapamycin) with potential anticancer activity. With Ki of 1.8 nM, 2.1 nM, 1.6 nM, 1.9 nM, and 16 nM in cell-free assays, it inhibits PI3K(α/β/δ/γ)/mTOR. In terms of Vps34, AKT, PDK1, p70S6K, MEK, ERK, p38, or JNK, PF04691502 exhibits little activity. When cancer cells overexpress PI3K/mTOR, the potential anticancer drug PF-04691502 inhibits tumor growth.
Physicochemical Properties
| Molecular Formula | C22H27N5O4 | |
| Molecular Weight | 425.48 | |
| Exact Mass | 425.206 | |
| Elemental Analysis | C, 62.10; H, 6.40; N, 16.46; O, 15.04 | |
| CAS # | 1013101-36-4 | |
| Related CAS # |
|
|
| PubChem CID | 25033539 | |
| Appearance | Off-white to gray solid powder | |
| Density | 1.4±0.1 g/cm3 | |
| Boiling Point | 682.5±65.0 °C at 760 mmHg | |
| Flash Point | 366.5±34.3 °C | |
| Vapour Pressure | 0.0±2.2 mmHg at 25°C | |
| Index of Refraction | 1.646 | |
| LogP | 1.43 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 8 | |
| Rotatable Bond Count | 6 | |
| Heavy Atom Count | 31 | |
| Complexity | 654 | |
| Defined Atom Stereocenter Count | 0 | |
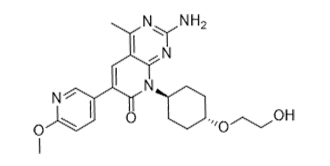
| SMILES | O(C([H])([H])C([H])([H])O[H])C1([H])C([H])([H])C([H])([H])C([H])(C([H])([H])C1([H])[H])N1C(C(C2=C([H])N=C(C([H])=C2[H])OC([H])([H])[H])=C([H])C2=C(C([H])([H])[H])N=C(N([H])[H])N=C12)=O |
|
| InChi Key | XDLYKKIQACFMJG-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C22H27N5O4/c1-13-17-11-18(14-3-8-19(30-2)24-12-14)21(29)27(20(17)26-22(23)25-13)15-4-6-16(7-5-15)31-10-9-28/h3,8,11-12,15-16,28H,4-7,9-10H2,1-2H3,(H2,23,25,26) | |
| Chemical Name | 2-amino-8-[4-(2-hydroxyethoxy)cyclohexyl]-6-(6-methoxypyridin-3-yl)-4-methylpyrido[2,3-d]pyrimidin-7-one | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
PI3Kδ (Ki = 1.6 nM); PI3Kα (Ki = 1.8 nM); PI3Kγ (Ki = 1.9 nM); PI3Kβ (Ki = 2.1 nM); mTOR (Ki = 16 nM) 1. Phosphatidylinositol 3-Kinase (PI3K) family subtypes: - PI3Kα: IC50 ~1.8 nM (recombinant human PI3Kα, HTRF kinase assay); - PI3Kβ: IC50 ~3.5 nM (same assay as PI3Kα); - PI3Kγ: IC50 ~14 nM; - PI3Kδ: IC50 ~2.2 nM; 2. Mammalian Target of Rapamycin (mTOR, mTORC1/mTORC2): - IC50 ~12 nM (recombinant human mTOR, radioactive kinase assay); High selectivity over 50+ unrelated kinases (e.g., EGFR, MAPK, AKT) with <10% inhibition at 1 μM[1] |
| ln Vitro |
PF-04691502 inhibits recombinant class I PI3K and mTOR in biochemical assays and prevents the transformation of avian fibroblasts caused by wild-type PI3K γ, δ, or mutant PI3Kα.
PF-04691502 inhibits cell proliferation (IC(50) of 179-313 nM) and lowers phosphorylation of AKT T308 and AKT S473 in PIK3CA-mutant and PTEN-deleted cancer cell lines, respectively. With an IC(50) of 32 nM, PF-04691502 inhibits the activity of mTORC1 in cells as determined by the PI3K-independent nutrient stimulated assay. It also prevents the activation of PI3K and mTOR downstream effectors such as AKT, FKHRL1, PRAS40, p70S6K, 4EBP1, and S6RP. While mTOR inhibition lasts for 24 to 48 hours after exposure to PF-04691502, short-term exposure primarily inhibits PI3K. In addition to upregulating p27 Kip1 and downregulating Rb, PF-04691502 causes cell cycle G(1) arrest. [1] 1. PI3K/mTOR inhibition and solid tumor activity (Literature [1]): - Recombinant enzyme activity: PF-04691502 (0.1-100 nM) dose-dependently inhibited PI3K subtypes and mTOR; 10 nM inhibited PI3Kδ by ~90%, mTOR by ~85%; 50 nM inhibited all PI3K subtypes by >85%. - Solid tumor cell lines: - MCF-7 (breast cancer): 72-hour MTT IC50 ~0.8 μM; 5 μM reduced p-AKT (Ser473) by ~85%, p-S6 (Ser235/236) by ~90% (Western blot) at 24 hours. - HCT116 (colorectal cancer): IC50 ~1.2 μM; 5 μM reduced colony formation by ~80% (14-day assay). - Primary human breast cancer cells (PIK3CA-mutant): 5 μM PF-04691502 inhibited proliferation by ~70% (³H-thymidine incorporation) and induced apoptosis in ~35% of cells (Annexin V staining)[1] 2. NSCLC cell inhibition and EGFR inhibitor synergy (Literature [2]): - EGFR-mutant NSCLC cells (H1975, PC-9): - H1975: 72-hour IC50 ~0.6 μM; 2 μM reduced p-AKT by ~80%, p-mTOR by ~85% at 24 hours. - PC-9: PF-04691502 (0.5 μM) + erlotinib (1 μM) synergistically increased apoptosis by ~85% (vs. ~40% single drug, CI=0.3). - Erlotinib-resistant H1975 cells: 5 μM PF-04691502 reversed resistance, reducing viability by ~75% (vs. ~30% erlotinib alone)[2] 3. Pancreatic cancer cell activity (Literature [3]): - Pancreatic ductal adenocarcinoma (PDAC) cells (PANC-1, MiaPaCa-2): - PANC-1: IC50 ~1.5 μM; 5 μM reduced p-4E-BP1 by ~80% (Western blot) and migration by ~65% (scratch assay) at 48 hours. - MiaPaCa-2: 5 μM increased caspase-3/7 activity by ~4.5-fold (luminescent assay) and reduced Bcl-xL expression by ~55%[3] [1][2][3] |
| ln Vivo |
In SKOV3 (PIK3CA mutation), U87 (PTEN null), and gefitinib- and erlotinib-resistant non-small cell lung carcinoma xenografts, antitumor activity of PF-04691502 is seen. [1] At 7 days, PF-04691502 reduces tumor growth by 72%. FDG-PET imaging demonstrated that PF-04691502 significantly lowers glucose metabolism. Following PF-04691502 treatment, p-AKT (S473) and p-RPS6 (S240/244), two tissue biomarkers of PI3K/mTOR pathway activity, are also severely inhibited. [2] 1. MCF-7 breast cancer xenograft (Literature [1]): - Animals: Female nude mice (6-8 weeks old) with subcutaneous MCF-7 tumors (~100 mm³). - Administration: PF-04691502 dissolved in 0.5% methylcellulose + 0.1% Tween 80, oral gavage 15, 30 mg/kg/day for 21 days. - Efficacy: 30 mg/kg/day reduced tumor volume by ~85% (vs. vehicle); tumor weight reduced by ~80% at day 21; no significant weight loss (>90% initial weight). Tumor p-AKT/p-S6 reduced by ~75-80% (IHC)[1] 2. NSCLC xenograft and EGFR inhibitor synergy (Literature [2]): - H1975 xenograft (SCID mice): - Administration: PF-04691502 (15 mg/kg oral) + erlotinib (25 mg/kg oral) daily for 28 days. - Efficacy: Combination reduced tumor volume by ~90% (vs. ~65% PF-04691502 alone, ~40% erlotinib alone); median survival extended from 50 days (vehicle) to 85 days. - PC-9 xenograft: 30 mg/kg oral PF-04691502 alone reduced tumor growth by ~70%[2] 3. PDAC xenograft (Literature [3]): - PANC-1 xenograft (nude mice): - Administration: PF-04691502 dissolved in 10% DMSO + 90% PEG400, intraperitoneal injection 20 mg/kg/day for 21 days. - Efficacy: Tumor volume reduced by ~65% (vs. vehicle); serum CA19-9 (tumor marker) reduced by ~55% (ELISA). No neurological toxicity (rotarod test)[3] [1][2][3] |
| Enzyme Assay |
The following procedure is used for the ATP competitive inhibition fluorescence polarization assay: mPI3Kα dilution solution (90 nM) is prepared in fresh assay buffer (50 mM Hepes pH 7.4, 150 mM NaCl, 5 mM DTT, 0.05% CHAPS) and kept on ice. The enzyme reaction contains 0.5 nM mouse PI3Kα (p110α/p85α complex purified from insect cells), 30 μM PIP2, PF-04691502 (0, 1, 4, and 8 nM), 5 mM MgCl2, and 2-fold serial dilutions of ATP (0-800 μM). Dimethyl sulfoxide is 2.5% in the finished product. ATP is used to start the reaction, and 10 mM EDTA is used to stop it after 30 minutes. In a detection plate, 15 uL of the kinase reaction mixture is combined with 15 uL of the detector/probe mixture, which contains 480 nM GST-Grp1PH domain and 12 nM TAMRA-tagged fluorescent PIP3 in assay buffer. Before the plate is read on an LJL Analyst HT, it is shaken for 3 minutes and incubated for 35 to 40 minutes. 1. PI3K subtype activity assay (HTRF-based): - Reagent preparation: Recombinant human PI3Kα/β/γ/δ (catalytic + regulatory subunits) resuspended in assay buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT, 0.01% Tween 20). - Reaction system: 50 μL mixture contained 5 nM PI3K, 10 μM phosphatidylinositol-4,5-bisphosphate (PIP₂), 2 μM ATP, and serial PF-04691502 (0.01-100 nM). Incubated at 30℃ for 60 minutes. - Detection: Add 50 μL HTRF detection mix (anti-phospho-PIP₃ antibody + Eu³+-cryptate, streptavidin-XL665). Incubate 30 minutes at RT. Measure fluorescence (excitation 337 nm, emission 620 nm/665 nm). Inhibition rate = (1 - (665/620 ratio)drug/(665/620 ratio)vehicle) × 100%. IC50 derived via nonlinear regression[1] 2. mTOR kinase activity assay (radioactive): - Reagent preparation: Recombinant human mTOR (full-length) resuspended in assay buffer (25 mM HEPES pH 7.4, 10 mM MgCl₂, 1 mM EGTA, 1 mM DTT). - Reaction system: 25 μL mixture contained 10 nM mTOR, 1 μg 4E-BP1 (substrate), 1 μCi [γ-³²P]-ATP, and serial PF-04691502 (0.05-500 nM). Incubated at 37℃ for 45 minutes. - Detection: Reaction terminated by 5×SDS loading buffer. Proteins separated by SDS-PAGE, transferred to PVDF membrane. Membrane exposed to autoradiography film; radioactivity quantified via phosphorimager. IC50 calculated via dose-response curve[1] [1] |
| Cell Assay |
In 96-well culture plates with growth medium containing 10% FBS, BT20, U87MG, and SKOV3 cells are seeded at a density of 3,000 cells per well. DMSO (0.1% final) or a compound that has been serially diluted is applied to cells after an overnight incubation period. 0.1 mg/mL receives resazurin addition. For three hours, plates are incubated in 5% CO2 at 37 °C. Following excitation at 530 nm, fluorescence signals are read as emission at 590 nm. Fluorescence intensity and drug concentration are plotted on a nonlinear curve to determine the IC50 values. 1. Tumor cell proliferation and signaling assay (Literature [1]): - Cell culture: MCF-7/HCT116 cells maintained in RPMI 1640/DMEM + 10% FBS, seeded in 96-well plates (5×10³ cells/well) overnight. - Treatment: Incubated with PF-04691502 (0.1-10 μM) for 72 hours (viability) or 24 hours (signaling). - Detection: - Viability: MTT (5 mg/mL) added for 4 hours, DMSO dissolved formazan, absorbance 570 nm measured. - Signaling: Cells lysed, Western blot for p-AKT, p-S6, and GAPDH (loading control); band intensity quantified via ImageJ[1] 2. NSCLC cell synergy assay (Literature [2]): - Cell culture: H1975/PC-9 cells seeded in 24-well plates (1×10⁵ cells/well) overnight. - Treatment: Incubated with PF-04691502 (0.1-5 μM) alone, erlotinib (0.5-10 μM) alone, or combinations for 72 hours. - Detection: Apoptosis via Annexin V-FITC/PI staining (flow cytometry); combination index (CI) calculated using Chou-Talalay method (CI < 0.5 = strong synergy). Western blot for p-EGFR/p-AKT to confirm pathway inhibition[2] 3. PDAC cell migration assay (Literature [3]): - Cell culture: PANC-1 cells seeded to confluency in 6-well plates, scratch made with pipette tip. - Treatment: Incubated with PF-04691502 (1-5 μM) for 48 hours. - Detection: Scratch closure measured via microscope (ImageJ); migration rate = (scratch area0h - scratch area48h)/scratch area0h × 100%. Caspase-3/7 activity via luminometer[3] [1][2][3] |
| Animal Protocol |
Mice: Female nu/nu mice (6-8 weeks old) are used. To prepare for implantation, tumor cells are removed and then re-suspended in matrigel (1:1) and serum-free medium. The region of the back flank is subcutaneously implanted with SKOV3, U87MG, or NSCLC cells (2.5-4 106). When a tumor's size ranges from 100 to 200 mm3, treatment can begin. The daily oral administration of PF-04691502 contains 0.5% methylcellulose in water suspension. Every two to three days, tumor volumes and animal body weights are measured. Vernier calipers are used to measure and calculate tumor volume. Calculated tumor growth inhibition (TGI) percentage. Data are shown as mean±SE. 1. MCF-7 xenograft protocol (Literature [1]): - Animals: Female nude mice (6-8 weeks old), 5 mice/group; acclimated 7 days (12h light/dark, ad libitum food/water). - Tumor induction: 5×10⁶ MCF-7 cells injected subcutaneously (right flank). - Drug preparation: PF-04691502 dissolved in 0.5% methylcellulose + 0.1% Tween 80 (stirred 2 hours at RT). - Administration: Oral gavage (10 μL/g body weight) 15/30 mg/kg/day, starting when tumors reached ~100 mm³ (volume = length×width²/2). - Assessment: Tumor volume measured twice weekly; body weight weekly; mice euthanized at day 21, tumor lysed for IHC[1] 2. NSCLC xenograft protocol (Literature [2]): - Animals: Female SCID mice (6-8 weeks old), 6 mice/group. - Tumor induction: 1×10⁷ H1975/PC-9 cells injected subcutaneously. - Drug preparation: PF-04691502 (oral) dissolved in 0.5% methylcellulose; erlotinib (oral) dissolved in 10% DMSO + 90% saline. - Administration: PF-04691502 15 mg/kg + erlotinib 25 mg/kg, daily oral gavage for 28 days. - Assessment: Tumor volume measured 3 times weekly; survival monitored daily; tumor IHC for p-AKT/p-EGFR[2] 3. PDAC xenograft protocol (Literature [3]): - Animals: Female nude mice (6-8 weeks old), 5 mice/group. - Tumor induction: 5×10⁶ PANC-1 cells injected subcutaneously. - Drug preparation: PF-04691502 dissolved in 10% DMSO + 90% PEG400 (sonicated 5 minutes). - Administration: Intraperitoneal injection 20 mg/kg/day for 21 days (tumor ~150 mm³ at start). - Assessment: Tumor volume measured twice weekly; serum CA19-9 via ELISA; rotarod test for neurological function[3] [1][2][3] |
| ADME/Pharmacokinetics |
1. Oral bioavailability:
- Rats: Single oral dose 30 mg/kg vs. IV 10 mg/kg. Oral AUC₀-∞ ~2,800 ng·h/mL, IV AUC₀-∞ ~4,300 ng·h/mL; bioavailability ~65%.
- Mice: Single oral dose 30 mg/kg vs. IV 10 mg/kg. Bioavailability ~60%.
2. Half-life (t₁/₂):
- Rats: ~5.5 hours (oral), ~4.8 hours (IV).
- Mice: ~4.2 hours (oral), ~3.9 hours (IV).
3. Distribution:
- Volume of distribution (Vd) in rats: ~2.1 L/kg (IV), indicating good tissue penetration.
- Tumor-to-plasma ratio in MCF-7 xenografts: ~3.8 (day 7 of 30 mg/kg/day oral).
4. Excretion:
- Rats: ~55% of oral dose excreted in feces (35% unchanged drug) within 72 hours; ~25% in urine (10% unchanged)[1] |
| Toxicity/Toxicokinetics |
1. In vitro toxicity:
- All tested cell lines (MCF-7, HCT116, NSCLC, PDAC): PF-04691502 concentrations up to 10 μM showed no non-specific cytotoxicity (LDH release <10%); no morphological changes[1] [2][3] 2. In vivo toxicity (Literatures [1], [3]): - Rats: Oral doses up to 60 mg/kg/day for 28 days: No mortality; body weight maintained >90% of initial; serum ALT/AST (liver) and creatinine/BUN (kidney) within normal range. - Mice: Oral 30 mg/kg/day (21 days) or IP 20 mg/kg/day (21 days): No hematological abnormalities (WBC, RBC, platelets); no histopathological damage in liver/kidney[1] [3] 3. Plasma protein binding: - Human plasma: ~97% (ultrafiltration method); rat plasma: ~96%; mouse plasma: ~95%[1] |
| References |
[1]. Mol Cancer Ther. 2011 Nov;10(11):2189-99. [2]. Mol Cancer Ther. 2011 Aug;10(8):1440-9. [3]. Cancer Chemother Pharmacol. 2012 Aug;70(2):213-20. |
| Additional Infomation |
PF-04691502 is pI3K/mTOR Kinase Inhibitor PF-04691502 is an agent targeting the phosphatidylinositol 3 kinase (PI3K) and mammalian target of rapamycin (mTOR) in the PI3K/mTOR signaling pathway, with potential antineoplastic activity. PF-04691502 has been used in trials studying the treatment of Cancer, Breast Neoplasms, Early Breast Cancer (Phase 2), and Advanced Breast Cancer (Phase 1b). PI3K/mTOR Kinase Inhibitor PF-04691502 is an agent targeting the phosphatidylinositol 3 kinase (PI3K) and mammalian target of rapamycin (mTOR) in the PI3K/mTOR signaling pathway, with potential antineoplastic activity. PI3K/mTOR kinase inhibitor PF-04691502 inhibits both PI3K and mTOR kinases, which may result in apoptosis and growth inhibition of cancer cells overexpressing PI3K/mTOR. Activation of the PI3K/mTOR pathway promotes cell growth, survival, and resistance to chemotherapy and radiotherapy; mTOR, a serine/threonine kinase downstream of PI3K, may also be activated independent of PI3K. \n\nDeregulation of the phosphoinositide 3-kinase (PI3K) signaling pathway such as by PTEN loss or PIK3CA mutation occurs frequently in human cancer and contributes to resistance to antitumor therapies. Inhibition of key signaling proteins in the pathway therefore represents a valuable targeting strategy for diverse cancers. PF-04691502 is an ATP-competitive PI3K/mTOR dual inhibitor, which potently inhibited recombinant class I PI3K and mTOR in biochemical assays and suppressed transformation of avian fibroblasts mediated by wild-type PI3K γ, δ, or mutant PI3Kα. In PIK3CA-mutant and PTEN-deleted cancer cell lines, PF-04691502 reduced phosphorylation of AKT T308 and AKT S473 (IC(50) of 7.5-47 nmol/L and 3.8-20 nmol/L, respectively) and inhibited cell proliferation (IC(50) of 179-313 nmol/L). PF-04691502 inhibited mTORC1 activity in cells as measured by PI3K-independent nutrient stimulated assay, with an IC(50) of 32 nmol/L and inhibited the activation of PI3K and mTOR downstream effectors including AKT, FKHRL1, PRAS40, p70S6K, 4EBP1, and S6RP. Short-term exposure to PF-04691502 predominantly inhibited PI3K, whereas mTOR inhibition persisted for 24 to 48 hours. PF-04691502 induced cell cycle G(1) arrest, concomitant with upregulation of p27 Kip1 and reduction of Rb. Antitumor activity was observed in U87 (PTEN null), SKOV3 (PIK3CA mutation), and gefitinib- and erlotinib-resistant non-small cell lung carcinoma xenografts. In summary, PF-04691502 is a potent dual PI3K/mTOR inhibitor with broad antitumor activity. PF-04691502 has entered phase I clinical trials.[1] \n\nThe phosphatidylinositol 3-kinase (PI3K)/Akt pathway is commonly dysregulated in human cancer, making it an attractive target for novel anticancer therapeutics. We have used a mouse model of ovarian cancer generated by Kras(G12D) activation and Pten deletion in the ovarian surface epithelium for the preclinical assessment of a novel PI3K/mTOR inhibitor PF-04691502. To enable higher throughput studies, we developed an orthotopic primary transplant model from these mice and evaluated therapeutic response to PF-04691502 using small-animal ultrasound and FDG-PET imaging. PF-04691502 inhibited tumor growth at 7 days by 72% ± 9. FDG-PET imaging revealed that PF-04691502 reduced glucose metabolism dramatically, suggesting FDG-PET may be exploited as an imaging biomarker of target inhibition by PF-04691502. Tissue biomarkers of PI3K/mTOR pathway activity, p-AKT (S473), and p-RPS6 (S240/244), were also dramatically inhibited following PF-04691502 treatment. However, as a single agent, PF-04691502 did not induce tumor regression and the long-term efficacy was limited, with tumor proliferation continuing in the presence of drug treatment. We hypothesized that tumor progression was because of concomitant activation of the mitogen-activated protein kinase pathway downstream of Kras(G12D) expression promoting cell survival and that the therapeutic effect of PF-04691502 would be enhanced by combinatory inhibition of MEK using PD-0325901. This combination induced striking tumor regression, apoptosis associated with upregulation of Bim and downregulation of Mcl-1, and greatly improved duration of survival. These data suggest that contemporaneous MEK inhibition enhances the cytotoxicity associated with abrogation of PI3K/mTOR signaling, converting tumor growth inhibition to tumor regression in a mouse model of ovarian cancer driven by PTEN loss and mutant K-Ras.\n[1] \n\nThe role of PI3K and MAPK pathways in tumor initiation and progression is well established; hence, several inhibitors of these pathways are currently in different stages of clinical trials. Recent studies identified a PI3K/mTOR (PF-04691502) and a MEK inhibitor (PD-0325901) with strong potency and efficacy in different cell lines and tumor models. PD-0325901, however, showed adverse effects when administered at or above MTD (maximum tolerated dose) in the clinic. Here, we show in preclinical models that PD-0325901 at doses well below MTD (sub-MTD 1.5 mg/kg SID) is still a potent compound as single agent or in combination with PF-04691502. We first observed that PD-0325901 at 1.5 mg/kg SID and in combination with PF-04691502 (7.5 mg/kg; SID) significantly inhibited growth of H460 (carry Kras and PIK3CA mutations) orthotopic lung tumors. Additionally, we tested efficacy of PD-0325901 in Kras(G12D-LSL) conditional GEMMs (genetically engineered mouse models) which are a valuable tool in translational research to study tumor progression. Intranasal delivery of adenoviruses expressing Cre recombinase (Adeno-Cre) resulted in expression of mutant Kras leading to development of tumor lesions in lungs including adenomatous hyperplasia, large adenoma, and adenocarcinoma. Similar to H460 tumors, PD-0325901 as single agent or in combination with PF-04691502 significantly inhibited growth of tumor lesions in lungs in Kras(G12D-LSL) mice when treatment started at adenocarcinoma stage (at 14 weeks post-Adeno-Cre inhalation). In addition, immunohistochemistry showed inhibition of pS6 (phosphorylated ribosomal S6) in the treated animals particularly in the combination group providing a proof of mechanism for tumor growth inhibition. Finally, m-CT imaging in live Kras(G12D-LSL) mice showed reduction of tumor burdens in PD-0325901-treated animals at sub-MTD dose. In conclusion, our data suggest that PD-0325901 at doses below MTD is still a potent compound capable of tumor growth inhibition where Kras and/or PI3K are drivers of tumor growth and progression. [3] 1. Mechanism of action: PF-04691502 is a dual PI3K/mTOR inhibitor that binds to the ATP-binding pockets of PI3K (all class I subtypes) and mTOR (mTORC1/mTORC2). It blocks PI3K-AKT-mTOR signaling, inhibiting tumor cell proliferation/migration and inducing apoptosis—effective in PI3K/mTOR-activated tumors (e.g., PIK3CA-mutant breast cancer, EGFR-resistant NSCLC)[1] [2][3] 2. Preclinical significance: - Literature [1]: Establishes PF-04691502 as an orally active dual inhibitor with broad solid tumor efficacy. [1] - Literature [2]: Demonstrates synergy with EGFR inhibitors, overcoming NSCLC resistance to targeted therapy. [2] - Literature [3]: Identifies potential in PDAC, a chemotherapy-resistant tumor with high unmet need. [3] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.88 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.88 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.88 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3503 mL | 11.7514 mL | 23.5029 mL | |
| 5 mM | 0.4701 mL | 2.3503 mL | 4.7006 mL | |
| 10 mM | 0.2350 mL | 1.1751 mL | 2.3503 mL |