PF-01247324 is a novel, potent, selective and orally bioavailable Nav1.8 channel blocker with an IC50 of 196 nM for recombinant human Nav1.8 channel. PF-01247324 was administered by oral gavage at 1000 mg/kg; control groups received an equal volume of vehicle. Behavioral assays of motor coordination, grip strength, and ataxia were performed. We observed significant improvements in motor coordination and cerebellar-like symptoms in mice that received PF-01247324 compared to control littermates that received vehicle. These preclinical proof-of-concept data suggest that PF-01247324, its derivatives, or other Nav1.8-selective blockers merit further study for providing symptomatic therapy for cerebellar dysfunction in MS and related disorders. NaV 1.8 ion channels have been highlighted as important molecular targets for the design of low MW blockers for the treatment of chronic pain.
Physicochemical Properties
| Molecular Formula | C13H10N3OCL3 |
| Molecular Weight | 330.597 |
| Exact Mass | 328.989 |
| CAS # | 875051-72-2 |
| PubChem CID | 11659955 |
| Appearance | Off-white to light yellow solid powder |
| LogP | 3.971 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 3 |
| Rotatable Bond Count | 2 |
| Heavy Atom Count | 20 |
| Complexity | 358 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | HPIUHDCRVYDAEJ-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C13H10Cl3N3O/c1-18-13(20)10-3-2-7(12(17)19-10)8-4-6(14)5-9(15)11(8)16/h2-5H,1H3,(H2,17,19)(H,18,20) |
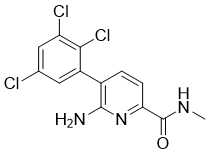
| Chemical Name | 6-amino-N-methyl-5-(2,3,5-trichlorophenyl)pyridine-2-carboxamide |
| Synonyms | PF-01247324; PF 01247324; PF01247324. |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | In human dorsal root ganglion (DRG) neurons, PF-01247324 inhibits both native tetrodotoxin-resistant (TTX-R) currents (IC50=331 nM) and recombinantly expressed hNav1.8 channels (IC50=196 nM). The selectivity of PF-01247324 is 50 times higher than that of the recombinantly expressed TTX-R hNav1.5 channel (IC50=10 μM), and 65–100 times higher than that of the TTX-sensitive (TTX-S) channel (IC50=10–18 μM). PF-01247324 was shown to alter the action potential waveform and decrease excitability in both rat and human DRG neurons by in vitro current clamp [1]. |
| ln Vitro |
In human dorsal root ganglion (DRG) neurons, PF-01247324 inhibits both native tetrodotoxin-resistant (TTX-R) currents (IC50=331 nM) and recombinantly expressed hNav1.8 channels (IC50=196 nM). The selectivity of PF-01247324 is 50 times higher than that of the recombinantly expressed TTX-R hNav1.5 channel (IC50=10 μM), and 65–100 times higher than that of the TTX-sensitive (TTX-S) channel (IC50=10–18 μM). PF-01247324 was shown to alter the action potential waveform and decrease excitability in both rat and human DRG neurons by in vitro current clamp [1]. PF-01247324 inhibited recombinant human Nav1.8 channels expressed in HEK293 cells with an IC50 of 196 nM, and native TTX-R currents in human DRG neurons with an IC50 of 331 nM. The inhibition was both state-dependent (shifting steady-state inactivation curves) and frequency-dependent (increased block with repetitive stimulation). In current-clamp recordings, 1 μM PF-01247324 significantly reduced evoked action potential firing in both rat and human DRG neurons and altered action potential waveform (reduced peak amplitude and depolarizing slope). In the presence of the inflammatory mediator PGE2, higher concentrations of PF-01247324 were required to achieve similar reductions in neuronal excitability. [1] |
| ln Vivo |
Its effectiveness in neuropathic pain and inflammatory models was shown in rodent trials. At 100 mg/kg, PF-01247324 decreased phase 2 withdrawal by 37%. PF-01247324, at a dose of 30 mg/kg, significantly mitigates the effects of carrageenan-induced heat hyperalgesia and CFA-induced mechanical hyperalgesia in rat models [1]. When PF-01247324 was administered to mice instead of controls, there were notable improvements in motor coordination and cerebellar-like symptoms [2]. Oral administration of PF-01247324 produced antinociceptive effects in multiple rodent pain models. In the rat formalin test, 100 mg/kg significantly reduced Phase 2 flinching by 37%. In the carrageenan-induced thermal hyperalgesia model, 30 mg/kg significantly increased paw withdrawal latency. In the CFA-induced mechanical hyperalgesia model, 30 mg/kg significantly increased mechanical threshold. In the spinal nerve ligation (SNL) neuropathic pain model, 10 and 30 mg/kg significantly reduced tactile allodynia at 1 and 2 hours post-dose. |
| Cell Assay |
Manual patch-clamp electrophysiology on recombinant channels: HEK293 cells stably expressing human Nav1.8 or other Nav isoforms were used. Cells were perfused with extracellular solution containing 132 mM NaCl, 5.4 mM KCl, 10 mM HEPES, 5 mM glucose, 1.8 mM CaCl2, and 0.8 mM MgCl2 (pH 7.4). For Nav1.5, sodium was partially replaced with choline chloride. Pipettes were filled with intracellular solution containing 110 mM CsF, 35 mM CsCl, 5 mM NaCl, 10 mM HEPES, and 10 mM EGTA (pH 7.3). Currents were evoked using voltage protocols from a holding potential of -120 mV (or -150 mV for Nav1.5) to 0 mV after an 8-second inactivating prepulse. Concentration-response curves were generated by applying increasing concentrations of PF-01247324. [1] PatchXpress automated electrophysiology: Used for site-directed mutagenesis studies on hNav1.8 channels (WT and F1710A/Y1717A mutant). Cells were harvested by trypsinization. The extracellular solution contained 135 mM NaCl, 2 mM CaCl2, 5.4 mM KCl, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.4). The intracellular solution contained 135 mM CsF, 5 mM NaCl, 2 mM MgCl2, 10 mM EGTA, and 10 mM HEPES (pH 7.4). Steady-state inactivation curves were run before each compound addition. [1] |
| Animal Protocol |
Rat formalin model: Male Sprague Dawley rats (170–300 g) were orally administered PF-01247324 (0, 10, 30, 100 mg/kg) in 0.5% methylcellulose/0.1% Tween 80 vehicle 4 hours before a subcutaneous injection of 2.5% formalin (50 μL) into the hind paw. Flinching behavior was automatically recorded for 60 minutes. [1] Rat carrageenan thermal hyperalgesia: Inflammation was induced by injecting 50 μL of 1% λ-carrageenan into the rat hind paw. PF-01247324 or vehicle was administered orally 1 hour after carrageenan, and thermal paw withdrawal latency was measured 2 hours later using a Hargreaves apparatus. [1] Rat CFA tactile allodynia: Inflammation was induced by injecting 150 μL of CFA emulsion into the hind paw 48 hours before testing. PF-01247324 or vehicle was administered orally, and mechanical thresholds were measured 4 hours later using von Frey filaments. [1] Rat spinal nerve ligation (SNL) model: Neuropathy was induced by ligating the L5 spinal nerve. Rats displaying baseline allodynia (threshold ≤6.6 g) were orally administered PF-01247324 (10 or 30 mg/kg), vehicle, or gabapentin (100 mg/kg) 1–2 hours before mechanical threshold testing. [1] Pharmacokinetic study in rats: Male Sprague-Dawley rats received a single intravenous bolus of 2 mg/kg or an oral dose of 5 mg/kg PF-01247324 formulated in 5% Tween 80 in glucose solution. Blood samples were collected serially via a vena cava cannula over 24 hours for plasma concentration analysis. [1] |
| ADME/Pharmacokinetics |
Following intravenous administration (2 mg/kg) in rats, systemic blood clearance (CL) was 11.7 mL/min/kg, and volume of distribution at steady state (Vss) was 3.0 L/kg. Following oral administration (5 mg/kg), the maximum plasma concentration (Cmax) was 474 ng/mL (50 nM unbound), and oral bioavailability (F) was 88%. The fraction absorbed (Fa) was estimated to be 1.0. PF-01247324 penetrated the central nervous system in rats, with an unbound brain-to-plasma concentration ratio of 1.5. [1] |
| Toxicity/Toxicokinetics |
In a functional observation battery (FOB) in rats, oral doses up to 600 mg/kg (unbound plasma concentration ~1.01 μM) showed no significant differences from vehicle in general behavior, motor function, sensory reactivity, or physiological parameters. In a mouse rotarod assay, doses up to 1000 mg/kg (unbound plasma concentration ~1.22 μM) did not impair motor coordination or balance. No overt toxicity or adverse phenotypes were reported in rats or mice at the tested doses. Cardiovascular effects were not assessed. [1] |
| References |
[1]. A novel selective and orally bioavailable Nav 1.8 channel blocker, PF-01247324, attenuates nociception and sensory neuron excitability. Br J Pharmacol. 2015 May;172(10):2654-70. [2]. Oral administration of PF-01247324, a subtype-selective Nav1.8 blocker, reverses cerebellar deficits in a mouse model of multiple sclerosis. PLoS One. 2015 Mar 6;10(3):e0119067. |
| Additional Infomation |
PF-01247324 (6-amino-5-(2,3,5-trichloro-phenyl)-pyridine-2-carboxylic acid methylamide) is a structurally novel, orally bioavailable, and state- and frequency-dependent Nav1.8 channel blocker. It represents a second-generation tool compound with high selectivity over other Nav subtypes and a different off-target profile compared to earlier blockers like A-803467. The study confirms the role of Nav1.8 channels in both inflammatory and neuropathic pain and demonstrates the compound's utility in reducing sensory neuron excitability in both rodent and human DRG neurons. [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ≥ 30 mg/mL (~90.74 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (7.56 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (7.56 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (7.56 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.0248 mL | 15.1240 mL | 30.2480 mL | |
| 5 mM | 0.6050 mL | 3.0248 mL | 6.0496 mL | |
| 10 mM | 0.3025 mL | 1.5124 mL | 3.0248 mL |