Physicochemical Properties
| Molecular Formula | C24H20CLN5O |
| Molecular Weight | 429.90 |
| Exact Mass | 429.135 |
| CAS # | 404844-11-7 |
| PubChem CID | 10181075 |
| Appearance | Solid powder |
| Density | 1.3±0.1 g/cm3 |
| Melting Point | 300ºC |
| Index of Refraction | 1.691 |
| LogP | 4.04 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 31 |
| Complexity | 570 |
| Defined Atom Stereocenter Count | 0 |
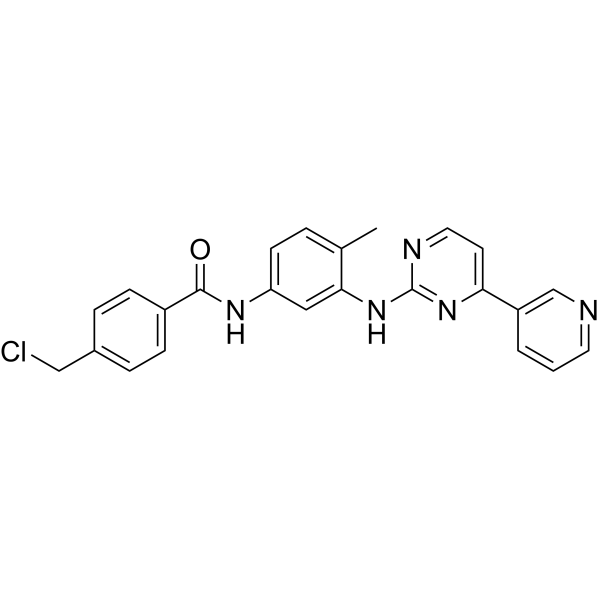
| SMILES | O=C(NC1=CC=C(C)C(NC2=NC=CC(C3=CC=CN=C3)=N2)=C1)C4=CC=C(CCl)C=C4 |
| InChi Key | WUGWDFMSJPJUEZ-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C24H20ClN5O/c1-16-4-9-20(28-23(31)18-7-5-17(14-25)6-8-18)13-22(16)30-24-27-12-10-21(29-24)19-3-2-11-26-15-19/h2-13,15H,14H2,1H3,(H,28,31)(H,27,29,30) |
| Chemical Name | 4-(chloromethyl)-N-[4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl]benzamide |
| Synonyms | 404844-11-7; 4-(Chloromethyl)-N-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)benzamide; 4-Chloromethyl-N-[4-methyl-3-[[4-(pyridin-3-yl)pyrimidin-2-yl]amino]phenyl]benzamide; N-[4-Methyl-3-(4-pyridin-3-yl-pyrimidin-2-ylamino)-phenyl]-4-chloromethyl Benzamide; 4-(chloromethyl)-N-[4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl]benzamide; PDGFR; A kinase inhibitor 2; CHEMBL1765715; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: This product requires protection from light (avoid light exposure) during transportation and storage. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Abl1 6.95 μM (IC50); KIT 2.45 μM (IC50); PDGFR 1.39 μM (IC50) |
| ln Vitro | Aside from PDGFRα kinase inhibitor 2 (compound 1), all other derivatives lost their activity against ABL, presumably due to the loss of ionic interaction between the positively charged methylated nitrogen of the piperazine ring of imatinib (36) and the carbonyl groups of Ile 789 and His 790 (KIT kinase numbering). On the other hand, the compounds containing the different electrophilic traps retained their activity against KIT and PDGFR. To confirm the formation of a covalent adduct between the imatinib analogues and the γ-sulfur of the targeted cysteine residue, tryptic digests of the kinases incubated with the different inhibitors were analyzed by mass spectrometry (MS). Compound 3, bearing the chloroacetamide group, indeed led to the disappearance of the fragment containing the targeted cysteine residue and to the appearance of a new peak corresponding to the peptide−inhibitor adduct for both KIT and PDGFRα. Further sequencing by MS/MS fragmentation indicated the expected shift in molecular weight (450 amu) for the cysteine adduct having reacted with compound 3 (Figure 4D). Other compounds failed to show similar covalent adducts. This can be rationalized by a less optimal positioning of the electrophile (for example, the angle between compound 1 warhead and the sulfur atom is unfavorable for SN2 substitution; Figure 4C) or a lack of sensitivity in the mass spectrometry assay.[1] |
| Enzyme Assay |
Human Kinase Data Set[1] Sequences corresponding to the 491 kinase catalytic domains of the 478 human eukaryotic protein kinases and their sequence alignment file were retrieved from the published data generated by Manning et al. This alignment has been refined for the purpose of this study (available upon request). For each kinase, the corresponding crystal structures were downloaded (July 2010) from the Protein Data Bank via the BIRD system. The part of the workflow (Figure 3) explaining the data cleaning of all chains extracted from crystal structures, as well as the classification into conformations, is detailed in Leproult et al. (to be submitted). This led to a total of 1357 chains, of which 974, 250, and 133 chains were classified into active, C-helix-out, and DFG-out conformations, respectively. ATP Binding Site Pocket Detection[1] Each chain was prepared with the PDB2PQR program, which was efficient for automatically removing ligands, solvent, and ions. This was also used to add hydrogens to all atoms in a manner consistent with favorable hydrogen bonding. To automatically detect empty pockets inside the chains, the in-house Pck program was used. This program is dedicated to the geometric-based detection and characterization of pockets. This uses the alpha shape theory to represent the surface of proteins and then implements algorithms specific of some types of pockets. Among these algorithms, CAST was preferentially chosen due to its ability to detect partially buried pockets, as observed for the kinase ATP binding site. Because the classification into conformations required the superimposition of all chains (to be submitted, Leproult et al.), the pocket corresponding to the ATP binding site was located by testing whether it contains a virtually created 3D point placed in the ATP binding site nearby the hinge region. In the end, only 23 chains were reported as having no detected ATP binding site pocket. Further inspection of these chains revealed that most of them have either a small pocket or a side chain that blocks a part of the ATP binding site, resulting in an autoinhibition phenomenon. Amino Acid Selection Inside the ATP Binding Site[1] Among the amino acids having atoms participating in the detected ATP pocket, those having at least one atom of the side chain that participates in the pocket are selected for the next part of the workflow. For those that only have main chain atoms that participate in the binding site, the secondary structure information using DSSP program is taken into account. Indeed, if the amino acid is located on a flexible structural element, such as a loop or a turn, then the amino acid is selected. The only peculiar case concerns glycine amino acid when located on a nonflexible structural element. Because a glycine amino acid does not have a side chain, the two hydrogens attached to the Cα atom are investigated. If the one generating an R chirality when mutated to the side chain of a cysteine participates in the detected ATP binding site pocket, then the glycine is selected. Robust Amino Acid Positions and Propagation to All Human Kinases[1] Each previously selected amino acid is highlighted in the sequence of the corresponding kinase, inside the sequence alignment file. This gives access to an amino acid position. Once the amino acid positions have been obtained for all chains, the ones appearing in more than 30% of all chains, with respect to the conformational class, are considered as robust positions. This step avoids keeping rare positions located in abnormally extended ATP binding sites. Indeed, the detection of some large ATP binding sites by Pck was due to the proximity of the ATP binding site with other domains or the lack of structural elements forming the ATP binding site in the chain. Next, robust amino acid positions are propagated to all human kinases using the sequence alignment. This allows the inference of amino acids having a side chain subject to participation in the ATP binding site for every human kinase in every conformation. The robust cysteine positions are the focus of this study and can be visualized in 3D on the following website: http://lbgi.igbmc.fr/Kinatown. Inhibition IC50 for ABL1 wt, KIT wt, and PDGFRα wt Kinases[1] A radiometric protein kinase assay (33PanQinase Activity Assay) was used for measuring the kinase activity of ABL1 wt, KIT wt, and PDGFRα wt kinases (Proqinase, Freiburg, Germany). All kinase assays were performed in 96-well FlashPlates from Perkin-Elmer (Boston, MA) using 50 μL of assay buffer (60 mM HEPES-NaOH, pH 7.5, 3 mM MgCl2, 3 mM MnCl2, 3 μM Na3VO4, 1.2 mM DTT, 50 μg mL−1 PEG2000, and 1 μM [γ-33P]ATP), 20 ng of kinase, and a generic substrate (polyGluTyr for KIT and polyAlaGluLsTyr for ABL and PDGFRα) with 1% DMSO. The test compound concentration ranged from 20 μM to 0.1 nM (semilog dilution). The assays were performed by premixing the ATP solution with the test compound and addition of this solution to the kinase/substrate solution. After 60 min at 30 °C, the reaction was stopped with 50 μL of 2% H3PO4, plates were aspirated and treated with 200 μL of 0.9% NaCl (2×), and the level of 33P incorporation was determined with a microplate scintillation counter. Mass Spectrometry of KIT Kinase and PDGFRα Kinase Complexes[1] A 50 μM solution of each compound was incubated with 2 μg of each kinase in 60 mM Hepes-NaOH, pH 7.5 (3 mM MgCl2 and 3 Mm MnCl2), for 12 h. The proteins were then isolated by one-dimensional sodium dodecyl sulfate−polyacrylamide gel electrophoresis gel, subjected to in-gel trypsin digest, and analyzed by matrix-assisted laser desorption/ionization. As compound 2 cannot form a covalent adduct, this sample was used as a negative control. Only compound 3 showed a new peak corresponding to the expected molecular weight from the peptide adduct (peptide containing Cys 466, MW = 1092.487 for recombinant KIT Kinase, and Cys 491 for recombinant PDGFRα kinase, MW = 1078.472). To further confirm the identity of the peptide sequence of the new peak, MS/MS experiments were performed, which indeed provide the expected degradation product corresponding to the following sequences: KIT = NC(compound 3)IHR; PDGFRα = NC(compound 3)VHR. Kinase Selectivity Profile[1] The selectivity profile was measured by using the KinomeScan technology (http://www.kinomescan.com/) based on active site-dependent competition binding assays. The results are expressed as a percentage of signal between a negative (DMSO) and a positive (known binder) control. Residual binding (%) = [(tested compound − positive control)/(tested compound − negative control) × 100]. |
| References |
[1]. Cysteine mapping in conformationally distinct kinase nucleotide binding sites: application to the design of selective covalent inhibitors. J Med Chem. 2011 Mar 10;54(5):1347-55. |
| Additional Infomation | Kinases have emerged as one of the most prolific therapeutic targets. An important criterion in the therapeutic success of inhibitors targeting the nucleotide binding pocket of kinases is the inhibitor residence time. Recently, covalent kinase inhibitors have attracted attention since they confer terminal inhibition and should thus be more effective than reversible inhibitors with transient inhibition. The most robust approach to design irreversible inhibitors is to capitalize on the nucleophilicity of a cysteine thiol group present in the target protein. Herein, we report a systematic analysis of cysteine residues present in the nucleotide binding site of kinases, which could be harnessed for irreversible inhibition, taking into consideration the different kinase conformations. We demonstrate the predictive power of this analysis with the design and validation of an irreversible inhibitor of KIT/PDGFR kinases. This is the first example of a covalent kinase inhibitor that combines a pharmacophore addressing the DFG-out conformation with a covalent trap. |
Solubility Data
| Solubility (In Vitro) | DMSO : 25 mg/mL (58.15 mM; with sonication (<60°C)) |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3261 mL | 11.6306 mL | 23.2612 mL | |
| 5 mM | 0.4652 mL | 2.3261 mL | 4.6522 mL | |
| 10 mM | 0.2326 mL | 1.1631 mL | 2.3261 mL |