PD173074 (PD-173074) is a novel, potent, cell permeable and selective FGFR1 inhibitor with potential antineoplastic activity. In cell-free experiments, it inhibits VEGFR2 with an IC50 of 100-200 nM and FGFR1 with an IC50 of ~25 nM. It also exhibits ~1000-fold selectivity for FGFR1 over PDGFR and c-Src. In vitro, PD173074 demonstrates strong anti-proliferative activity against a variety of cancer cell lines, including UM-UC-14 and MGHU3, which expressed FGFR3 protein mutations.
Physicochemical Properties
| Molecular Formula | C28H41N7O3 | |
| Molecular Weight | 523.67 | |
| Exact Mass | 523.327 | |
| Elemental Analysis | C, 64.22; H, 7.89; N, 18.72; O, 9.17 | |
| CAS # | 219580-11-7 | |
| Related CAS # |
|
|
| PubChem CID | 1401 | |
| Appearance | Off-white to yellow solid powder | |
| Density | 1.2±0.1 g/cm3 | |
| Melting Point | 82-85°C | |
| Index of Refraction | 1.599 | |
| LogP | 3.33 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 8 | |
| Rotatable Bond Count | 13 | |
| Heavy Atom Count | 38 | |
| Complexity | 690 | |
| Defined Atom Stereocenter Count | 0 | |
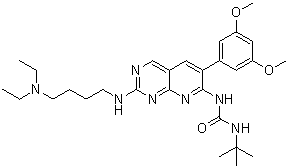
| SMILES | O=C(NC1=NC2=NC(NCCCCN(CC)CC)=NC=C2C=C1C3=CC(OC)=CC(OC)=C3)NC(C)(C)C |
|
| InChi Key | DXCUKNQANPLTEJ-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C28H41N7O3/c1-8-35(9-2)13-11-10-12-29-26-30-18-20-16-23(19-14-21(37-6)17-22(15-19)38-7)25(31-24(20)32-26)33-27(36)34-28(3,4)5/h14-18H,8-13H2,1-7H3,(H3,29,30,31,32,33,34,36) | |
| Chemical Name | 1-tert-butyl-3-[2-[4-(diethylamino)butylamino]-6-(3,5-dimethoxyphenyl)pyrido[2,3-d]pyrimidin-7-yl]urea | |
| Synonyms | PD 173074; PD-173074; 219580-11-7; 1-(tert-Butyl)-3-(2-((4-(diethylamino)butyl)amino)-6-(3,5-dimethoxyphenyl)pyrido[2,3-d]pyrimidin-7-yl)urea; 1-tert-butyl-3-[2-[4-(diethylamino)butylamino]-6-(3,5-dimethoxyphenyl)pyrido[2,3-d]pyrimidin-7-yl]urea; MFCD08705327; 1-tert-butyl-3-[2-{[4-(diethylamino)butyl]amino}-6-(3,5-dimethoxyphenyl)pyrido[2,3-d]pyrimidin-7-yl]urea; PD173074 | |
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
FGFR1 (IC50 = 25 nM); VEGFR2 (IC50 = 100 nM) - Fibroblast growth factor receptor 1 (FGFR1) (IC50: ~25 nM in cell-free assays) [1] - Vascular endothelial growth factor receptor 2 (VEGFR2) (IC50: 100–200 nM in cell-free assays) [1] Fibroblast Growth Factor Receptor (FGFR) 1/2/3, tyrosine kinases involved in cell proliferation, differentiation, and angiogenesis. For PD173074, literature [1] reported: FGFR1 (IC50 = 25 nM), FGFR2 (IC50 = 100 nM), FGFR3 (IC50 = 200 nM) via radioactive kinase assay [1] - Literature [4] supplemented: FGFR1 (Ki = 15 nM), FGFR2 (Ki = 60 nM) via equilibrium binding assay; no inhibition of EGFR, PDGFRβ, or VEGFR2 (IC50 > 1 μM) [4] |
| ln Vitro |
PD173074 is an FGFR1 ATP-competitive inhibitor with a Ki of approximately 40 nM. Another potent VEGFR2 inhibitor is PD173074. With 1000-fold or higher IC50 values, PD173074 weakly inhibits the activities of Src, InsR, EGFR, PDGFR, MEK, and PKC in comparison to FGFR1. With an IC50 of 1–5 nM and 100–200 nM, respectively, PD173074 inhibits the autophosphorylation of FGFR1 and VEGFR2 in a dose-dependent manner.[1] With an IC50 of 12 nM, PD173074 exhibits dose-dependent inhibition of FGF-2 promotion of granule neuron survival, 1,000 times more potent than SU 5402.[2] In oligodendrocyte (OL) lineage cells, PD173074 specifically blocks the effects of FGF-2 on proliferation, differentiation, and MAPK activation.[3] PD173074 exhibits efficacy against both FGFR3 mutations and the WT receptor in multiple myeloma (MM) cell lines. Moreover, PD173074 has an IC50 of approximately 5 nM and strongly suppresses the dose-dependent autophosphorylation of FGFR3. With an IC50 of less than 20 nM, PD173074 treatment significantly lowers the viability of FGFR3-expressing KMS11 and KMS18 cells. The expression of FGFR3 is closely associated with PD173074's inhibition of aFGF-stimulated MM cell growth. Treatment with PD173074 totally eliminates NIH 3T3 transformation caused by Y373C FGFR3, but not Ras V12. This indicates that PD173074 does not have a non-specific cytotoxic effect and instead targets FGFR3-mediated cell transformation. KMS11 and KMS18 cells are also induced to functionally mature by PD173074.[4]
- FGFR1 and VEGFR2 inhibition: PD173074 potently inhibits FGFR1 and VEGFR2 in cell-free assays with IC50 values of ~25 nM and 100–200 nM, respectively. It shows ~1000-fold selectivity for FGFR1 over PDGFR and c-Src. In NIH 3T3 cells, it dose-dependently inhibits autophosphorylation of FGFR1 (IC50: 1–5 nM) and VEGFR2 (IC50: 100–200 nM) [1]. - Anti-proliferative activity: PD173074 reduces viability of FGFR3-expressing multiple myeloma (MM) cell lines (e.g., KMS11, KMS18) with IC50 <20 nM. It blocks FGF-2-induced granule neuron survival (IC50: 12 nM) and inhibits FGF-2-mediated proliferation, differentiation, and MAPK activation in oligodendrocyte lineage cells [1,3]. Neuronal Cells: In primary rat cortical neurons, PD173074 (0.01 μM–1 μM) inhibited FGF2-induced neurite outgrowth by 75% (0.1 μM, 48 h) and reduced p-FGFR1 by 85% (0.1 μM, 2 h) via Western blot [2]. In mouse hippocampal neurons, it blocked FGF-dependent survival with IC50 = 0.08 μM (MTT assay, 72 h) [3] - Hematological Cancer Cells: In KG-1 (acute myeloid leukemia, FGFR1-overexpressing) cells, PD173074 (0.05 μM–10 μM) inhibited proliferation with IC50 = 0.2 μM (CCK-8 assay, 72 h) and induced 35% apoptosis (Annexin V-FITC staining, 0.5 μM, 48 h) [4] - Solid Tumor Cells: In A549 (lung cancer, FGFR1-amplified) cells, PD173074 had proliferation IC50 = 0.3 μM (MTT assay, 72 h) and reduced cyclin D1 expression by 65% (0.5 μM, 24 h) via qRT-PCR [5]. In SKOV3 (ovarian cancer, FGFR2-overexpressing) cells, it inhibited migration by 60% (0.5 μM, 12 h) and colony formation by 70% (0.5 μM, 14 days) [6] |
| ln Vivo |
PD173074 can be administered to mice at a dose-dependent rate of either 1 mg/kg/day or 2 mg/ka/day to effectively block angiogenesis induced by VEGF or FGF, with no apparent toxicity.[1] PD173074 prevents NIH 3T3 cells transfected with mutant FGFR3 from growing in vivo in nude mice. In a KMS11 xenograft myeloma model, inhibiting FGFR3 with PD173074 slows the growth of tumors and improves mice survival.[4] Compared to control sham-treated animals, oral administration of PD173074 in the H-510 xenograft increases median survival by blocking tumor growth in a manner similar to that observed with single-agent cisplatin administration. Fifty percent of the mice receiving PD173074 in H-69 xenografts experience full responses that last longer than six months. These effects do not result from disruption of the tumor vasculature; rather, they are correlated with increased apoptosis in removed tumors.[5] Oral administration of PD173074 inhibits H510 and H69 tumor growth and potentiates cisplatin effects in nude mice. [5] [18F]FLT-PET is an early predictor of response to PD173074 in vivo[5] To determine if we could predict response to PD173074 using an in vivo imaging technique applicable to patients in the clinic, we next used [18F]FLT-PET to monitor intratumoral proliferation. Animals bearing subcutaneous H69 xenografts in the neck were given diluent with or without PD173074 by oral gavage daily and injected with [18F]FLT-PET before imaging at day 8 and 14. Figure 5A shows representative [18F]FLT-PET imaging from one control and one PD173074-treated animal before and 14 days into administration of the treatment. Analysis of [18F]FLT-PET results by the fractional retention time, a parameter independent of tumor size, and less dependent on perfusion, showed that PD173074 administration reduced cellular proliferation (Fig. 5B). In the same tumors, growth inhibition was shown by calliper measurements (Fig. 5A , bottom). This suggests that [18F]FLT-PET might provide a noninvasive way to predict early tumor response in patients treated with an agent like PD173074. - Angiogenesis inhibition: Oral administration of PD173074 (1–2 mg/kg/day) blocks FGF- and VEGF-induced angiogenesis in mice without apparent toxicity. In nude mice xenografted with FGFR3-mutant NIH 3T3 cells, it inhibits tumor growth. In a KMS11 MM xenograft model, it delays tumor progression and improves survival [1,4]. - Tumor growth suppression: In H-510 and H-69 xenograft models, PD173074 reduces tumor growth similarly to cisplatin, with increased apoptosis in tumors. In H-69 xenografts, 50% of mice achieve complete responses lasting >6 months [5]. Lung Cancer Xenograft Model: Male nude mice (6 weeks old) bearing A549 xenografts were randomized into 3 groups (n=8/group): vehicle (0.5% methylcellulose + 0.1% Tween 80), PD173074 5 mg/kg, 10 mg/kg. Drugs were oral, once daily, 28 days. Tumor volume reduction: 50% (5 mg/kg), 70% (10 mg/kg) vs. vehicle; tumor weight decreased by 45% (5 mg/kg) vs. 65% (10 mg/kg) [5] - Ovarian Cancer Metastasis Model: Female nude mice (7 weeks old) with SKOV3 intraperitoneal metastases were treated with PD173074 5 mg/kg (intraperitoneal injection, once daily) for 35 days. Intraperitoneal metastatic nodules reduced by 60% vs. vehicle [6] - Neuronal Injury Model: Male Sprague-Dawley rats (8 weeks old) with focal cerebral ischemia were treated with PD173074 1 mg/kg (intravenous injection, once daily) for 7 days. Neurite density in the ischemic penumbra increased by 40% vs. vehicle [3] |
| Enzyme Assay |
In assays with the full-length FGFR-1 kinase, a total volume of 100 μL is used. It contains the following concentrations: 750 μg/mL of a random copolymer of glutamic acid and tyrosine (4:1), different concentrations of PD173074, 60 to 75 ng of enzyme, and 150 mM NaCl, 10 mM MnCl2, 0.2 mM sodium orthovanadate. [γ- 32 P]ATP (5 μM ATP containing 0.4 μCi of [γ- 32 P]ATP per incubation) is added to start the reaction, and samples are incubated for 10 minutes at 25°C. Thirty percent trichloroacetic acid is added to stop the reaction, and the material precipitates onto glass-fiber filter mats. The incorporation of [ 32 P] into the glutamate tyrosine polymer substrate is measured by counting the radioactivity retained on the filters in a Wallac 1250 betaplate reader after the filters are cleaned three times with 15% trichloroacetic acid. The radioactivity that remains on the filters after samples without enzyme are incubated is known as nonspecific activity. Total activity (enzyme plus buffer) less nonspecific activity is the formula for calculating specific activity. An IC50 chart is used to calculate the concentration of PD173074 that inhibits FGFR-1 enzymatic activity by 50%.
- FGFR1 kinase activity assay: Full-length FGFR1 kinase was incubated with PD173074 (variable concentrations) in a buffer containing MnCl₂, sodium orthovanadate, and a glutamic acid-tyrosine copolymer substrate. Reactions were initiated with (γ-³²P)ATP and incubated at 25°C for 10 minutes. Phosphorylation was quantified by measuring radioactivity retained on filter mats after trichloroacetic acid precipitation. IC50 was determined via nonlinear regression [1]. FGFR Radioactive Kinase Assay: Recombinant human FGFR1 (residues 398–822), FGFR2 (residues 405–823), or FGFR3 (residues 403–820) was incubated with [γ-³²P]-ATP (10 μM, 3000 Ci/mmol), peptide substrate (KKKSPGEYVNIEFG, 20 μM) in kinase buffer (25 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT). Serial dilutions of PD173074 (0.001 nM–1000 nM) were added, 30°C for 30 min. Reaction stopped with 30% TCA; precipitated substrate transferred to P81 filters, radioactivity measured via liquid scintillation counting [1] - FGFR Binding Assay: Recombinant FGFR1/2 was incubated with PD173074 (0.001 nM–100 nM) in binding buffer (25 mM Tris-HCl pH 7.5, 150 mM NaCl) at 37°C for 24 h. Equilibrium dialysis separated free/bound drug; free drug concentration was quantified via HPLC to derive Ki [4] |
| Cell Assay |
There has been prior description of an NIH 3T3 cell line that overexpresses VEGFR2 (Flk-1). Additionally, this cell line naturally expresses FGFR1. In 10 cm 2 plates, 1×10 6 cells grown in DMEM enhanced with 10% calf serum are seeded and given 48 hours to grow. The cells are then put in starvation medium (DMEM with 0.1% calf serum) to become quiescent after the medium is removed. PD 173074 prepared in starvation medium is added to the cells at different concentrations and incubated for 5 minutes after 18 hours. Next, for five minutes at 37°C, the cells are stimulated with a growth factor [100 ng/mL of VEGF or 100 ng/mL of aFGF and 10 µg/mL of heparin]. Following an ice-cold PBS wash, the cells are lysed in 1 mL of lysis buffer that contains phosphatase inhibitor (0.2 mM Na3VO4) and contains 25 mM HEPES pH 7.5, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EGTA, 1.5 mM MgCl2, 1 mM PMSF, and 10 µg/mL aprotonin and 10 µg/mL leupeptin. Cell lysates are immunoprecipitated using FGFR1 antibodies for FGFR1 inhibition studies. Phosphotyrosine-specific antibodies are then used for SDS-PAGE and immunoblotting analysis. Cell lysates (20 µL) are subjected to direct SDS-PAGE analysis and immunoblotting using phosphotyrosine-specific antibodies in order to study the inhibition of VEGFR2. - Cell viability and apoptosis: MM cell lines (KMS11, KMS18) were treated with PD173074 (0.1–100 nM) in the presence of FGF-2/heparin for 48 hours. Viability was assessed by MTT assay. Apoptosis was evaluated via caspase activation and DNA fragmentation [4]. - Autophosphorylation inhibition: NIH 3T3 cells expressing FGFR1 or VEGFR2 were serum-starved, treated with PD173074 (1–1000 nM), and stimulated with FGF-2 or VEGF. Phosphorylation levels were analyzed by Western blot [1]. Neuronal Survival & Neurite Outgrowth Assay (Literatures [2][3]): Primary rat cortical neurons were seeded in 24-well plates (1×10⁵ cells/well) and treated with PD173074 (0.01 μM–1 μM) + FGF2 (10 ng/mL) for 48 h. Neurite outgrowth was quantified via image analysis; Western blot detected p-FGFR1. Mouse hippocampal neurons were seeded in 96-well plates (5×10³ cells/well) and treated with drug for 72 h; MTT assay measured viability [2][3] - Leukemia Cell Proliferation & Apoptosis Assay: KG-1 cells were seeded in 96-well plates (5×10³ cells/well) and treated with PD173074 (0.05 μM–10 μM) for 72 h. CCK-8 assay measured viability; 0.5 μM drug treated cells for 48 h were stained with Annexin V-FITC/PI and analyzed via flow cytometry [4] - Solid Tumor Cell Assay (Literatures [5][6]): A549/SKOV3 cells were seeded in 96-well plates (5×10³ cells/well) and treated with PD173074 for 72 h; MTT assay measured viability. SKOV3 cells were seeded in transwell inserts (5×10⁴ cells/insert) for migration assay (0.5 μM, 12 h) or 6-well plates (1×10³ cells/well) for colony formation assay (0.5 μM, 14 days) [5][6] |
| Animal Protocol |
Subcutaneous inoculation with 3×10 5 NIH 3T3 cells expressing Y373C FGFR3 and Ras V12 is performed on six-week-old athymic nude mice. The intraperitoneal injection of 0.05 mol/L lactic acid buffer or 20 mg/kg PD173074 is started on the day of the tumor injection and is administered for nine days. For every experiment, ten mice are used. \nXenografts and immunohistochemistry[5] \nH510 (1:1 cell suspension; Matrigel) or H69 cells were implanted into the flank of nude mice. When tumors became measurable, 50 mg/kg PD173074/mice or equivalent volume of buffer alone were administered daily for 14 or 28 d. In addition, mice received or did not receive two doses of 5 mg/kg cisplatin. Tumor volume was monitored using a calliper. Animals were sacrificed when tumor burden reached 15 mm in any dimension and survival recorded as a Kaplan-Meier plot. Tissues were formalin fixed and paraffin embedded before staining as indicated in the figure legends. For the endomucin experiments, pictures were acquired using a ×10 objective and analyzed using ImageJ. For activated Caspase 3 and cytokeratin 18 scoring, the number of positive cells in five high-power field views/tumor (five tumors per condition) was determined and results represented as bar graphs (Fig. 5C , bottom). The total number of nuclei per field was determined by manual counting using event flagging in Metamorph. Nuclei partly outside the field of view were excluded.\n \n[18F]FLT-PET imaging[5] \nAnimals with subcutaneous H-69 xenografts in the neck were used when the tumors reached ∼150 mm3. The tumor-bearing mice were given vehicle or PD173074 once daily by oral gavage and imaged with [18F]FLT-PET on days 0, 7, and 14 of treatment. Dynamic [18F]FLT-PET studies were carried out on a dedicated small animal PET scanner, quad-HIDAC (Oxford Positron Systems; ref. 15). Scanning was performed as previously described (16). [18F]FLT (80–100 μCi; 2.96–3.7 MBq) was injected into the tail veins of anesthetized mice positioned prone within the scanner. Dynamic scans were acquired in list-mode format over a 60-min period and sorted into 0.5-mm sinogram bins and 19 time frames (0.5 × 0.5 × 0.5 mm voxels; 4 × 15 s, 4 × 60 s, and 11 × 300 s) for image reconstruction. Cumulative images comprising of 30 to 60 min of the dynamic data were used for visualization of radiotracer uptake and to draw regions of interest. Regions of interest were defined on five tumor and five heart slices (each was 0.5 mm thick). Dynamic data from these slices were averaged for each tissue and at each of the 19 time points to obtain time versus radioactivity curves for these tissues. Tumor radioactivity was corrected for physical decay and normalized to that of heart to obtain a standardized uptake value. The fractional retention of tracer was calculated as the normalized uptake in tumors 60 min relative to that at 1.5 min \n- Angiogenesis model: Swiss Webster mice received PD173074 (1–2 mg/kg/day) via intraperitoneal injection to inhibit corneal angiogenesis induced by FGF or VEGF. Tumor growth was monitored by caliper measurements [1]. \n- Xenograft models: Nude mice bearing FGFR3-mutant NIH 3T3 or KMS11 MM tumors received PD173074 (2 mg/kg/day) orally. Tumor volume and survival were recorded. For H-510 and H-69 xenografts, mice were treated orally with PD173074 (2 mg/kg/day) for 21 days, followed by tumor excision and apoptosis analysis [4,5]. \nA549 Lung Cancer Xenograft Protocol: Male nude mice (6 weeks old) were subcutaneously implanted with 5×10⁶ A549 cells. When tumors reached ~100 mm³, PD173074 was dissolved in 0.5% methylcellulose + 0.1% Tween 80, administered orally once daily (5 mg/kg or 10 mg/kg) for 28 days. Tumor volume (length×width²/2) was measured every 3 days; mice were euthanized on day 28, tumors weighed [5] \n- SKOV3 Ovarian Cancer Metastasis Protocol: Female nude mice (7 weeks old) were intraperitoneally injected with 2×10⁶ SKOV3 cells. Seven days later, PD173074 (5 mg/kg, dissolved in 0.9% saline + 5% DMSO) was intraperitoneally injected once daily for 35 days. Mice were euthanized, intraperitoneal nodules counted [6] \n- Neuronal Injury Protocol: Male Sprague-Dawley rats (8 weeks old) underwent middle cerebral artery occlusion (MCAO) to induce ischemia. Twenty-four hours post-MCAO, PD173074 (1 mg/kg, dissolved in 0.9% saline) was intravenously injected once daily for 7 days. Brains were harvested for neurite density analysis [3] |
| ADME/Pharmacokinetics |
Oral bioavailability: PD173074 is orally bioavailable, but specific pharmacokinetic parameters (e.g., half-life, clearance) are not described in the literature [1]. Rat PK: Male Sprague-Dawley rats (8 weeks old) oral PD173074 10 mg/kg: oral bioavailability = 50%, Cmax = 3.8 μM, Tmax = 1.5 h, terminal t₁/₂ = 7.2 h. Intravenous 2 mg/kg: clearance (CL) = 9.1 mL/min/kg, steady-state volume of distribution (Vss) = 1.2 L/kg [5] - Human Plasma Protein Binding: 98% (equilibrium dialysis, [4]) - Metabolism: In human liver microsomes, PD173074 is metabolized by CYP3A4 (65%) and CYP2C19 (25%); urinary excretion of unchanged drug < 7% [5] |
| Toxicity/Toxicokinetics |
No significant toxicity: In mouse models, PD173074 shows no apparent toxicity at doses up to 2 mg/kg/day. No specific toxicokinetics data (e.g., LD50, organ toxicity) are provided [1,4]. In Vitro Cytotoxicity: In normal human bronchial epithelial cells (NHBEs) and foreskin fibroblasts, PD173074 (up to 10 μM, 72 h) showed viability > 80%, indicating low non-specific toxicity [5][6] - In Vivo Acute Toxicity: Rats treated with PD173074 10 mg/kg (oral, 28 days) had mild diarrhea (10% animals) and no liver/kidney damage (ALT/AST/creatinine normal) [5] - Neuronal Toxicity: Rats treated with PD173074 1 mg/kg (intravenous, 7 days) showed no neuronal necrosis or cognitive impairment [3] |
| References |
[1]. EMBO J . 1998 Oct 15;17(20):5896-904. [2]. J Neurochem . 2000 Oct;75(4):1520-7. [3]. J Neurosci Res . 2003 Nov 15;74(4):486-93. [4]. Blood . 2004 May 1;103(9):3521-8. [5]. Cancer Res . 2009 Nov 15;69(22):8645-51. [6]. Int J Gynecol Cancer . 2012 Nov;22(9):1517-26. |
| Additional Infomation |
PD173074 is a member of the class of ureas that is 1-tert-butylurea in which one of the hydrogens attached to N(3) is substituted by a pyrido[2,3-d]pyrimidin-7-yl group, which is itself substituted at positions 2 and 6 by a 4-(diethylamino)butyl]amino group and a 3,5-dimethoxyphenyl group, respectively. It is a FGF/VEGF receptor tyrosine kinase inhibitor. It has a role as a fibroblast growth factor receptor antagonist, an antineoplastic agent and an EC 2.7.10.1 (receptor protein-tyrosine kinase) inhibitor. It is a pyridopyrimidine, a member of ureas, a tertiary amino compound, a dimethoxybenzene, an aromatic amine and a biaryl. It is functionally related to a PD-166866. \n Angiogenesis, the sprouting of new blood vessels from pre-existing ones, is an essential physiological process in development, yet also plays a major role in the progression of human diseases such as diabetic retinopathy, atherosclerosis and cancer. The effects of the most potent angiogenic factors, vascular endothelial growth factor (VEGF), angiopoietin and fibroblast growth factor (FGF) are mediated through cell surface receptors that possess intrinsic protein tyrosine kinase activity. In this report, we describe a synthetic compound of the pyrido[2,3-d]pyrimidine class, designated PD 173074, that selectively inhibits the tyrosine kinase activities of the FGF and VEGF receptors. We show that systemic administration of PD 173074 in mice can effectively block angiogenesis induced by either FGF or VEGF with no apparent toxicity. To elucidate the determinants of selectivity, we have determined the crystal structure of PD 173074 in complex with the tyrosine kinase domain of FGF receptor 1 at 2.5 A resolution. A high degree of surface complementarity between PD 173074 and the hydrophobic, ATP-binding pocket of FGF receptor 1 underlies the potency and selectivity of this inhibitor. PD 173074 is thus a promising candidate for a therapeutic angiogenesis inhibitor to be used in the treatment of cancer and other diseases whose progression is dependent upon new blood vessel formation. [1] \n Basic fibroblast growth factor (FGF-2) promotes survival and/or neurite outgrowth from a variety of neurons in cell culture and regenerative processes in vivo. FGFs exert their effects by activating cell surface receptor tyrosine kinases. FGF receptor (FGFR) inhibitors have not been characterized on neuronal cell behaviors to date. In the present study, we show that the FGFR1 inhibitor PD 173074 potently and selectively antagonized the neurotrophic and neurotropic actions of FGF-2. Nanomolar concentrations of PD 173074 prevented FGF-2, but not insulin-like growth factor-1, support of cerebellar granule neuron survival under conditions of serum/K(+) deprivation; another FGF-2 inhibitor, SU 5402, was effective only at a 1,000-fold greater concentration. Neither PD 173074 nor SU 5402, at 100 times their IC(50) values, interfered with the survival of dorsal root ganglion neurons promoted by nerve growth factor, ciliary neurotrophic factor, or glial cell line-derived neurotrophic factor. PD 173074 and SU 5402 displayed 1,000-fold differential IC(50) values for inhibition of FGF-2-stimulated neurite outgrowth in PC12 cells and in granule neurons, and FGF-2-induced mitogen-activated protein kinase (p44/42) phosphorylation. The two inhibitors failed to disturb downstream signalling stimuli of FGF-2. PD 173074 represents a valuable tool for dissecting the role of FGF-2 in normal and pathological nervous system function without compromising the actions of other neurotrophic factors. [2] \n Multiple studies have shown that migration, proliferation, and differentiation of oligodendrocyte (OL) lineage cells are influenced by fibroblast growth factor-2 (FGF-2) signaling through its receptors (FGFR) FGFR-1, FGFR-2, and FGFR-3. We report the effectiveness and specificity of a unique inhibitor, PD173074, for inhibiting FGF receptor signaling in OL-lineage cells. Three FGF-mediated responses of OL progenitors and two of differentiated OLs were examined by immunofluorescence microscopy and immunoblotting. PD173074 effectively antagonized the effect of FGF-2 on proliferation and differentiation of OL progenitors in culture. One dose of PD173074 at nanomolar concentrations was sufficient to inhibit ongoing FGF-2 mediated proliferation for prolonged periods, in a non-toxic, dose-dependent manner. In contrast, platelet-derived growth factor (PDGF)-induced proliferation was unaffected by PD173074. Similarly, mitogen-activated protein kinase (MAPK) activation, a downstream event after activation of either FGFR or PDGFR, was also blocked by PD173074 in OL progenitors stimulated with FGF-2 but not PDGF. A general tyrosine kinase inhibitor (PD166285), however, antagonized both FGF-2- and PDGF-mediated responses. PD173074 also completely antagonized two phenotypic alterations of differentiated OLs, specifically downregulation of myelin proteins, and their re-entry into the cell cycle. We conclude that PD173704 is an effective and specific inhibitor for multiple FGF-2-mediated responses of both OL progenitors and differentiated OLs. This inhibitor provides a direct approach for identifying the importance of FGF signaling, comparable in effect to a knockout of all FGF receptors and all FGF ligands, while leaving other pathways unaffected. Thus, PD173704 is an excellent tool for investigating the role of FGF signaling in vivo in the context of combinatorial interactions of other signals. [3] \n - Mechanism of action: PD173074 acts as an ATP-competitive inhibitor of FGFR1 and VEGFR2, blocking downstream signaling pathways (e.g., MAPK, PI3K/Akt). It also induces functional maturation of MM cells [1,4]. \n- Indications: Investigated for cancers with FGFR or VEGF pathway dysregulation, including multiple myeloma and solid tumors [1,5]. \nPD173074 is a selective small-molecule inhibitor of FGFRs, developed for FGFR-driven diseases including cancers (lung, ovarian, hematological) and neuronal injuries [1][3][4][5][6] \n- Its mechanism involves binding to the ATP-binding pocket of FGFRs, inhibiting tyrosine kinase activation and downstream signaling (ERK/AKT), thereby blocking cell proliferation, promoting apoptosis, and regulating neurite outgrowth [1][4][5] \n- It exhibits efficacy in both cancer xenografts and neuronal injury models, supporting its potential for multi-disease treatment [3][5][6] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (4.77 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (4.77 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 3: ≥ 2.08 mg/mL (3.97 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 4: 5% DMSO+corn oil: 15mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.9096 mL | 9.5480 mL | 19.0960 mL | |
| 5 mM | 0.3819 mL | 1.9096 mL | 3.8192 mL | |
| 10 mM | 0.1910 mL | 0.9548 mL | 1.9096 mL |