Physicochemical Properties
| Molecular Formula | C24H29N7O |
| Molecular Weight | 431.533364057541 |
| Exact Mass | 431.243 |
| CAS # | 2299226-01-8 |
| PubChem CID | 138466007 |
| Appearance | Light yellow to yellow solid powder |
| Density | 1.23±0.1 g/cm3(Predicted) |
| LogP | 2.7 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 8 |
| Heavy Atom Count | 32 |
| Complexity | 615 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | LYVJDLHFTGYNAV-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C24H29N7O/c1-5-29(6-2)22-11-9-18(15-26-22)23-27-20-14-17(24(32)25-4)8-10-21(20)31(23)16-19-12-13-30(7-3)28-19/h8-15H,5-7,16H2,1-4H3,(H,25,32) |
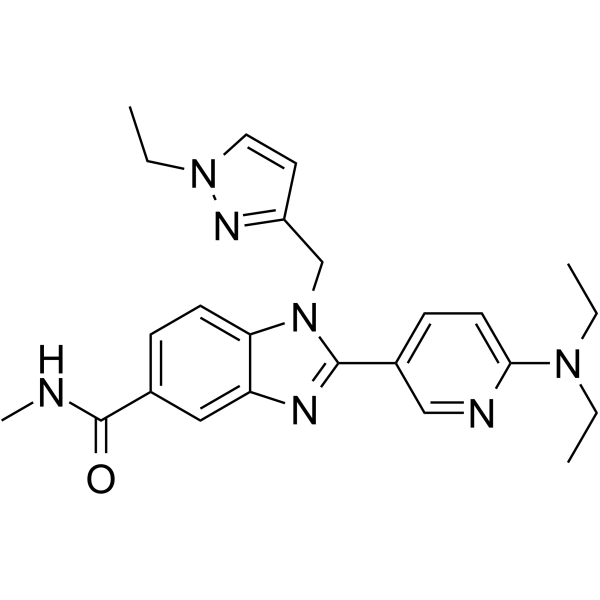
| Chemical Name | 2-[6-(diethylamino)pyridin-3-yl]-1-[(1-ethylpyrazol-3-yl)methyl]-N-methylbenzimidazole-5-carboxamide |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
P300/CBP-IN-3 targets p300 histone acetyltransferase (HAT) (IC50 = 0.18 μM) [1] P300/CBP-IN-3 targets CBP histone acetyltransferase (HAT) (IC50 = 0.25 μM) [1] P300/CBP-IN-3 targets c-MYC (inhibits c-MYC transcriptional activity with EC50 = 0.8 μM in c-MYC reporter assay) [1] |
| ln Vitro |
- p300/CBP HAT inhibitory activity: P300/CBP-IN-3 potently inhibited the acetyltransferase activity of recombinant human p300 and CBP HAT domains in a dose-dependent manner, with IC50 values of 0.18 μM and 0.25 μM respectively. It showed no significant inhibition against other histone acetyltransferases (GCN5, PCAF) at 10 μM [1] - Anti-proliferative activity: The compound inhibited the proliferation of various cancer cell lines dependent on p300/CBP or c-MYC. IC50 values were 1.2 μM (A549, lung cancer), 0.9 μM (MCF-7, breast cancer), 1.5 μM (HCT116, colon cancer), and 0.7 μM (NCI-H929, multiple myeloma). It had minimal cytotoxicity to normal human bronchial epithelial cells (BEAS-2B, IC50 > 20 μM) [1] - Inhibition of c-MYC transcriptional activity: In c-MYC luciferase reporter assay, P300/CBP-IN-3 (0.1-5 μM) dose-dependently suppressed c-MYC-mediated transcription, with EC50 = 0.8 μM. RT-PCR confirmed that it reduced mRNA levels of c-MYC target genes (Cyclin D1, c-Myc, Survivin) by 65%, 72%, and 58% respectively at 2 μM in A549 cells [1] - Reduction of histone acetylation: Western blot analysis showed that P300/CBP-IN-3 (0.5-2 μM) decreased acetylation levels of histone H3 (H3Ac) and histone H4 (H4Ac) in MCF-7 cells, with maximum inhibition of 70% (H3Ac) and 65% (H4Ac) at 2 μM. It did not affect total H3 or H4 protein levels [1] - Inhibition of clonogenicity: P300/CBP-IN-3 (0.3-1 μM) suppressed colony formation of A549 and NCI-H929 cells. At 1 μM, colony formation rates were reduced by 78% (A549) and 82% (NCI-H929) compared to control [1] - Induction of apoptosis: Flow cytometry analysis showed that P300/CBP-IN-3 (2 μM) induced apoptosis in NCI-H929 cells, with apoptotic rate increased from 5% (control) to 38%. Western blot detected increased cleavage of caspase-3 and PARP [1] |
| ln Vivo |
- Antitumor efficacy in xenograft models: In A549 lung cancer xenograft-bearing nude mice, oral administration of P300/CBP-IN-3 (15 mg/kg, 30 mg/kg, once daily for 21 days) resulted in tumor growth inhibition rates of 56% and 73% respectively. In NCI-H929 multiple myeloma xenografts, intraperitoneal administration of 20 mg/kg (once daily for 14 days) achieved a tumor growth inhibition rate of 68% [1] - Mechanism in vivo: Tumor tissues from treated mice (30 mg/kg, oral) showed reduced H3Ac and H4Ac levels (by 62% and 58% respectively) and downregulated expression of Cyclin D1 and Survivin (by 65% and 59% respectively) compared to vehicle control [1] - Tolerability: No significant body weight loss (< 8%) or obvious toxic signs (lethargy, diarrhea) were observed in treated mice at effective doses [1] |
| Enzyme Assay |
- p300/CBP HAT activity assay: Recombinant human p300 or CBP HAT domain was mixed with histone H3 substrate, acetyl-CoA, and P300/CBP-IN-3 at gradient concentrations (0.01-5 μM) in reaction buffer (pH 7.4). The mixture was incubated at 37°C for 1 hour, and acetylated histone H3 was detected by a colorimetric assay based on antibody recognition. IC50 values were calculated by plotting inhibition rate against drug concentration [1] - Histone acetyltransferase selectivity assay: Recombinant GCN5 and PCAF enzymes were separately mixed with their corresponding substrates, acetyl-CoA, and P300/CBP-IN-3 (10 μM) in reaction buffer. After 37°C incubation for 1 hour, enzyme activity was detected by colorimetric assay to evaluate selectivity [1] - c-MYC reporter assay: HEK293 cells stably transfected with c-MYC luciferase reporter plasmid were seeded into 96-well plates, treated with P300/CBP-IN-3 (0.1-5 μM) for 24 hours. Luciferase activity was measured and normalized to protein concentration to calculate EC50 for inhibiting c-MYC transcriptional activity [1] |
| Cell Assay |
- Cell viability assay: Cancer cells (A549, MCF-7, HCT116, NCI-H929) and BEAS-2B cells were seeded into 96-well plates at 4×10³ cells/well, treated with P300/CBP-IN-3 (0.05-20 μM) for 72 hours. Cell viability was measured by tetrazolium salt-based assay, and IC50 values were calculated [1] - Western blot assay: MCF-7 cells were treated with P300/CBP-IN-3 (0.5-2 μM) for 24 hours, lysed, and proteins were separated by SDS-PAGE. Membranes were probed with antibodies against H3Ac, H4Ac, total H3, total H4, cleaved caspase-3, cleaved PARP, and GAPDH. Band intensities were quantified by densitometry [1] - RT-PCR assay: A549 cells were treated with P300/CBP-IN-3 (0.5-2 μM) for 16 hours, total RNA was extracted and reverse-transcribed to cDNA. Quantitative PCR was performed to detect mRNA levels of Cyclin D1, c-Myc, and Survivin, with GAPDH as internal control [1] - Clonogenic assay: A549 and NCI-H929 cells were treated with P300/CBP-IN-3 (0.3-1 μM) for 24 hours, then seeded into 6-well plates at 400 cells/well and incubated for 14 days. Colonies were stained with crystal violet, counted, and inhibition rate was calculated relative to control [1] - Apoptosis assay: NCI-H929 cells were treated with P300/CBP-IN-3 (2 μM) for 48 hours, stained with Annexin V-FITC and propidium iodide (PI), and apoptotic cells were analyzed by flow cytometry [1] |
| Animal Protocol |
- A549 lung cancer xenograft model: Female nude mice (6-7 weeks old) were subcutaneously injected with A549 cells (4×10⁶ cells/mouse). When tumors reached ~120 mm³, mice were randomly divided into vehicle control, 15 mg/kg, and 30 mg/kg P300/CBP-IN-3 groups (n=7 per group). The compound was dissolved in a mixture of DMSO, PEG400, and sterile water (volume ratio 1:2:7) to prepare oral suspension, administered once daily for 21 days. Tumor volume was measured every 3 days, and body weight was recorded weekly [1] - NCI-H929 multiple myeloma xenograft model: Female nude mice were subcutaneously injected with NCI-H929 cells (5×10⁶ cells/mouse). When tumors reached ~100 mm³, mice were divided into vehicle control and 20 mg/kg P300/CBP-IN-3 groups (n=6 per group). The compound was dissolved in DMSO + PEG400 + sterile water (1:2:7) and administered intraperitoneally once daily for 14 days. Tumor volume and body weight were monitored as above [1] - Tumor tissue analysis: At the end of treatment, mice were sacrificed, tumors were excised, weighed, and stored at -80°C. Tumor lysates were used for Western blot analysis of H3Ac, H4Ac, Cyclin D1, and Survivin [1] |
| ADME/Pharmacokinetics |
- Plasma protein binding: P300/CBP-IN-3 had a plasma protein binding rate of 90.2 ± 2.1% in human plasma, determined by equilibrium dialysis [1] - In vitro metabolic stability: The compound showed good metabolic stability in human liver microsomes, with a half-life (t1/2) of 6.4 hours and metabolic clearance rate of 0.28 mL/min/mg protein [1] - In vivo pharmacokinetics in mice: After a single oral dose of 30 mg/kg, Cmax was 8.9 μM, AUC₀₋₂₄h was 56.3 μM·h, elimination half-life (t1/2) was 5.7 hours, and oral bioavailability (F) was 46.8%. After a single intraperitoneal dose of 20 mg/kg, Cmax was 11.2 μM, AUC₀₋₂₄h was 62.5 μM·h, and t1/2 was 6.1 hours [1] |
| Toxicity/Toxicokinetics |
- Acute toxicity: Mice showed no mortality or obvious toxicity symptoms after a single oral dose of P300/CBP-IN-3 up to 300 mg/kg, with maximum tolerated dose (MTD) > 300 mg/kg [1] - Subacute toxicity: In mice treated with P300/CBP-IN-3 (30 mg/kg, oral, once daily for 28 days), no significant changes were observed in body weight, blood routine parameters (WBC, RBC, PLT), or liver/kidney function indices (ALT, AST, creatinine, urea nitrogen). Histopathological examination of major organs (heart, liver, spleen, lungs, kidneys) revealed no abnormal lesions [1] |
| References |
[1]. 5-(1H-Benzo[d]imidazol-2-yl)pyridin-2-amine and 5-(3H-imidazo[4,5-b]pyridin-6-yl)pyridin-2-amine derivatives as c-MYC and p300/CBP histone acetyltransferase inhibitors for treating cancer and their preparation. |
| Additional Infomation |
- Chemical classification: P300/CBP-IN-3 is a small-molecule inhibitor, belonging to the 5-(1H-benzo[d]imidazol-2-yl)pyridin-2-amine or 5-(3H-imidazo[4,5-b]pyridin-6-yl)pyridin-2-amine derivative class [1] - Mechanism of action: The compound binds to the acetyl-CoA binding pocket of p300/CBP HAT domains, competitively inhibiting their acetyltransferase activity. This reduces histone acetylation, suppresses c-MYC transcriptional activity and expression of its target genes (Cyclin D1, Survivin), thereby inhibiting tumor cell proliferation and inducing apoptosis [1] - Target background: p300 and CBP are homologous histone acetyltransferases that regulate gene transcription by acetylating histones and transcription factors (e.g., c-MYC). Aberrant activation of p300/CBP and overexpression of c-MYC are associated with the development and progression of various cancers [1] - Therapeutic potential: P300/CBP-IN-3 is a potent, selective, and orally bioavailable p300/CBP and c-MYC inhibitor, showing therapeutic potential for the treatment of multiple cancers (e.g., lung cancer, breast cancer, colon cancer, multiple myeloma) [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~250 mg/mL (~579.33 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (4.82 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (4.82 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (4.82 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3173 mL | 11.5867 mL | 23.1734 mL | |
| 5 mM | 0.4635 mL | 2.3173 mL | 4.6347 mL | |
| 10 mM | 0.2317 mL | 1.1587 mL | 2.3173 mL |