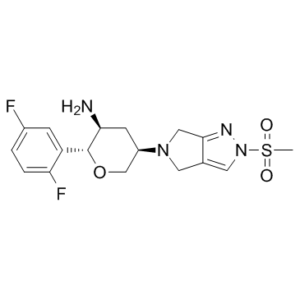
Omarigliptin (formerly known as MK-3102; MK3102) is a potent, selective, oral and long-acting DPP-4 (dipeptidyl peptidase 4) inhibitor with antidiabetic properties. At a 1.6 nM IC50, it inhibits DPP-4. Omarigliptin exhibits high selectivity (IC50 > 67 μM) against all proteases tested. At IKr, Caγ1.2, and Naγ1.5, its ion channel activity is weak (IC50 > 30 μmol/L). Furthermore, in every assay within an extensive selectivity counterscreen comprising 168 radioligand binding or enzymatic assays, an IC50 > 10 μmol/L was achieved. Under hyperglycemia, omarigliptin binds quickly and competitively to the DPP-4 active site. This process is highly selective and reversible, resulting in elevated insulin and decreased glucagon levels. It is presently undergoing a phase 3 clinical trial and has good pharmacokinetic profiles appropriate for once-weekly dosing.
Physicochemical Properties
| Molecular Formula | C17H20F2N4O3S | |
| Molecular Weight | 398.43 | |
| Exact Mass | 398.122 | |
| Elemental Analysis | C, 51.25; H, 5.06; F, 9.54; N, 14.06; O, 12.05; S, 8.05 | |
| CAS # | 1226781-44-7 | |
| Related CAS # |
|
|
| PubChem CID | 46209133 | |
| Appearance | White to off-white solid powder | |
| Density | 1.6±0.1 g/cm3 | |
| Boiling Point | 529.4±60.0 °C at 760 mmHg | |
| Flash Point | 274.0±32.9 °C | |
| Vapour Pressure | 0.0±1.4 mmHg at 25°C | |
| Index of Refraction | 1.689 | |
| LogP | 0.46 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 8 | |
| Rotatable Bond Count | 3 | |
| Heavy Atom Count | 27 | |
| Complexity | 649 | |
| Defined Atom Stereocenter Count | 3 | |
| SMILES | S(C([H])([H])[H])(N1C([H])=C2C(C([H])([H])N(C2([H])[H])[C@@]2([H])C([H])([H])O[C@]([H])(C3C([H])=C(C([H])=C([H])C=3F)F)[C@]([H])(C2([H])[H])N([H])[H])=N1)(=O)=O |
|
| InChi Key | MKMPWKUAHLTIBJ-ISTRZQFTSA-N | |
| InChi Code | InChI=1S/C17H20F2N4O3S/c1-27(24,25)23-7-10-6-22(8-16(10)21-23)12-5-15(20)17(26-9-12)13-4-11(18)2-3-14(13)19/h2-4,7,12,15,17H,5-6,8-9,20H2,1H3/t12-,15+,17-/m1/s1 | |
| Chemical Name | (2R,3S,5R)-2-(2,5-difluorophenyl)-5-(2-methylsulfonyl-4,6-dihydropyrrolo[3,4-c]pyrazol-5-yl)oxan-3-amine | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
DPP-4 (IC50 = 1.6 nM)
Dipeptidyl Peptidase 4 (DPP4) (IC50 = 1.6 nM for human recombinant DPP4; Ki = 0.6 nM) [1] - No significant inhibition of DPP8 (IC50 > 10 μM), DPP9 (IC50 > 10 μM), FAP (IC50 > 10 μM), or prolyl endopeptidase (PEP, IC50 > 10 μM), showing >6000-fold selectivity for DPP4 [1] |
| ln Vitro |
Omarigliptin is a potent DPP-4 inhibitor with weak ion channel activity (IC50 > 30 μmol/L at IKr, Caγ1.2, and Naγ1.5) and strong selectivity over other proteases tested (IC50 > 67 μmol/L). Furthermore, in every assay within an extensive selectivity counterscreen comprising 168 radioligand binding or enzymatic assays, an IC50 > 10 μmol/L was achieved. Under hyperglycemic circumstances, omagliptin binds quickly and competitively to the DPP-4 active site, a reversible and highly selective process that raises insulin levels and lowers glucagon levels[2]. Omarigliptin (0.01-100 nM) dose-dependently inhibited human recombinant DPP4 enzyme activity, with 95% inhibition at 10 nM [1] - The drug showed high species cross-reactivity: IC50 = 1.8 nM (rat DPP4), IC50 = 2.1 nM (dog DPP4), IC50 = 1.5 nM (monkey DPP4) [1] - Omarigliptin (1 nM) inhibited DPP4-mediated degradation of GLP-1 (7-36) amide by 90% in human plasma, prolonging the half-life of GLP-1 from 1.5 minutes to 35 minutes [1] - In Caco-2 cells expressing human DPP4, Omarigliptin (5 nM) reduced cell-surface DPP4 activity by 85% without affecting cell viability (>95% viability at 1 μM) [1] |
| ln Vivo |
In an oral glucose tolerance test (OGTT), it was given orally to lean mice one hour before the dextrose challenge. It significantly decreased blood glucose excursion in a dose-dependent manner, going from 0.01 mg/kg (7% reduction in glucose AUC) to 0.3 mg/kg (51% reduction). Plasma concentrations of active GLP-1 are dose-dependently increased upon omarigliptin administration. The male Sprague-Dawley rat and beagle dog exhibit low plasma clearance (0.9−1.1 mL/min/kg), 0.8−1.3 L/kg at steady state for the volume of distribution, and a long terminal half-life (∼11−22 h) in relation to the pharmacokinetics of omarigliptin. Omaligliptin has a good oral bioavailability in both dogs and rats (approximately 100%). Throughout the course of the trial, omajiptin is well tolerated; no death or adverse physical symptoms are observed[1]. After volunteers received a single oral dose of 25 mg, omarigliptin was absorbed quickly, reaching peak concentrations (Cmax) of 750 nmol/L in less than one hour (Tmax). The estimated bioavailability was 74%[2]. db/db mice (type 2 diabetes model) were administered Omarigliptin (0.1-10 mg/kg, oral gavage, once weekly for 4 weeks). Fasting blood glucose (FBG) was reduced by 40% (10 mg/kg) and glycated hemoglobin (HbA1c) by 0.8% compared to vehicle controls [1] - Omarigliptin (3 mg/kg, po, once weekly) in Zucker diabetic fatty (ZDF) rats reduced postprandial glucose AUC0-4h by 35% and increased plasma active GLP-1 levels by 2.8-fold [1] - In non-human primates (cynomolgus monkeys) with diet-induced hyperglycemia, Omarigliptin (1 mg/kg, po, once weekly) maintained FBG within normal range for 7 days, with HbA1c reduction of 0.6% after 8 weeks [2] - The drug did not cause hypoglycemia in normoglycemic mice or monkeys even at 30 mg/kg weekly dose [1][2] |
| Enzyme Assay |
Omarigliptin is a potent DPP-4 inhibitor with weak ion channel activity (IC50 > 30 μmol/L at IKr, Caγ1.2, and Naγ1.5) and strong selectivity over other proteases tested (IC50 > 67 μmol/L). Furthermore, in every assay within an extensive selectivity counterscreen comprising 168 radioligand binding or enzymatic assays, an IC50 > 10 μmol/L was achieved. Under hyperglycemic circumstances, omagliptin binds quickly and competitively to the DPP-4 active site, a reversible and highly selective process that raises insulin levels and lowers glucagon levels. In Vitro Pharmacology[1] Omarigliptin is a competitive, reversible inhibitor of DPP-4 (IC50 = 1.6 nM, Ki = 0.8 nM) and is more potent than sitagliptin (IC50 = 18 nM). It is highly selective over all proteases tested (IC50 > 67 μM), including QPP, FAP, PEP, DPP8, and DPP9. The compound has weak ion channel activity (IC50 > 30 μM at IKr, Cav1.2, and Nav1.5). An expansive selectivity counterscreen (168 radioligand binding or enzymatic assays) was carried out at MDS Pharma. An IC50 > 10 μM was obtained in all assays. DPP4 enzyme activity assay: Recombinant human DPP4 (50 pM) was incubated with fluorogenic substrate (H-Ala-Pro-AMC, 100 μM) in reaction buffer (pH 7.4) at 37°C. Serial concentrations of Omarigliptin (0.001-100 nM) were added, and the mixture was incubated for 30 minutes. Fluorescence intensity (excitation/emission = 360/460 nm) of cleaved AMC was measured, and IC50/Ki values were calculated by nonlinear regression [1] - DPP family selectivity assay: Recombinant DPP8, DPP9, FAP, and PEP (50 pM each) were incubated with respective fluorogenic substrates and Omarigliptin (0.01-10 μM) under optimized conditions. Enzyme activity was quantified to confirm DPP4-specific inhibition [1] |
| Cell Assay |
Cell-surface DPP4 inhibition assay: Caco-2 cells were cultured in DMEM medium supplemented with fetal bovine serum until confluent. Cells were treated with Omarigliptin (0.1-100 nM) for 2 hours, then incubated with H-Ala-Pro-AMC substrate (100 μM) at 37°C. Fluorescence from cell lysates was measured to assess residual DPP4 activity [1] - GLP-1 stability assay: Human plasma was mixed with GLP-1 (7-36) amide (100 nM) and Omarigliptin (0.1-10 nM), incubated at 37°C for 60 minutes. Active GLP-1 levels were quantified by ELISA to evaluate degradation inhibition [1] |
| Animal Protocol |
12 weeks, C57BL/6 male mice 2.5, 5 mg/kg P.o.; once a week for 8 weeks (50 mg/kg streptozotocin (STZ); i.p.; daily for five days) In Vivo Pharmacology in Preclinical Species[1] Omarigliptin was evaluated for its ability to improve glucose tolerance in lean mice. When orally administered 1 h prior to dextrose challenge in an oral glucose tolerance test (OGTT), it significantly reduced blood glucose excursion in a dose-dependent manner from 0.01 mg/kg (7% reduction in glucose AUC) to 0.3 mg/kg (51% reduction). The efficacy of glucose lowering in this model was similar to that achieved with sitagliptin. In the corresponding pharmacodynamic (PD) assay, omarigliptin-mediated plasma DPP-4 inhibition and plasma compound concentrations were dose-dependent. At the 0.3 mg/kg dose (corresponding to maximum acute glucose lowering efficacy), plasma DPP-4 activity was inhibited by 85% (uncorrected for assay dilution), which exceeds the target inhibition (80%) associated with maximal glucose lowering efficacy. The observed plasma DPP-4 inhibition was consistent with the measured plasma inhibitor concentration (521 nM) and the potency of the compound against murine plasma DPP-4 (IC50 = 43.9 nM in 50% mouse plasma). In addition, the administration of omarigliptin dose-dependently increased plasma concentrations of active GLP-1 (GLP-1[7–36]amide and GLP-1[7–37]) in this study, with the maximal increase in active GLP-1 observed at the 0.3–1 mg/kg dosages. The augmentation of active GLP-1 levels achieved at these doses (>10-fold) was in the range of elevation in circulating hormone observed in DPP-4-deficient (Dpp4–/–) mice (3- to 8-fold) relative to wild type animals db/db mouse type 2 diabetes model: 8-week-old male db/db mice were randomly divided into control (vehicle) and Omarigliptin groups (0.1, 1, 10 mg/kg). The drug was dissolved in 0.5% carboxymethylcellulose sodium, administered via oral gavage once weekly for 4 weeks. FBG was measured weekly; HbA1c was analyzed at endpoint. Plasma active GLP-1 and insulin levels were quantified by ELISA [1] - ZDF rat model: 10-week-old male ZDF rats were treated with Omarigliptin (3 mg/kg, po, once weekly) or vehicle for 6 weeks. Postprandial glucose was monitored at 0, 1, 2, 4 hours after glucose challenge; plasma metabolic markers were measured at endpoint [1] - Cynomolgus monkey model: Adult cynomolgus monkeys with diet-induced hyperglycemia were administered Omarigliptin (1 mg/kg, po, once weekly) for 8 weeks. FBG and HbA1c were measured every 2 weeks; safety parameters (hematology, clinical chemistry) were monitored throughout [2] |
| ADME/Pharmacokinetics |
Pharmacokinetics (PK) in Preclinical Species[1] PK experiments were generally conducted as follows: All species were fasted overnight before dosing, provided water ad libitum, and fed 4 h following drug treatment. Blood was collected at predetermined intervals for all species into EDTA-containing tubes and centrifuged. Plasma was harvested and stored at −70 °C until analysis. Test compounds were typically formulated as solutions in saline. Fasted male Sprague–Dawley rats were given either an iv dose of test compound solution via a cannula implanted in the femoral vein (n = 2) or a po dose by gavage (n = 3). Serial blood samples were collected at 5 (iv only), 15, and 30 min and at 1, 2, 4, 6, 8, 24, and 48 h postdose. Plasma was collected by centrifugation, and plasma concentrations of test compound were determined by LC–MS/MS following protein precipitation with acetonitrile. Fasted dogs were administered intravenous doses via the cephalic vein (dogs, n = 2). Oral doses were administered via gastric gavage (n = 2). Serial blood samples were collected at 5 (iv only), 15, and 30 min and at 1, 2, 4, 6, 8, 24, 30, 48, and 72 h postdose. Plasma was collected by centrifugation, and plasma concentrations of test compound were determined by LC–MS/MS following protein precipitation with acetonitrile. Pharmacokinetic parameters were calculated by established noncompartmental methods. The pharmacokinetics of omarigliptin in male Sprague–Dawley rat and beagle dog were characterized by a low plasma clearance (0.9–1.1 mL min–1 kg–1), a volume of distribution at steady state of 0.8–1.3 L/kg, and a long terminal half-life (∼11–22 h) (Table 1). The oral bioavailability of omarigliptin was good in both dogs and rats (∼100%). The mean percentage of unbound [3H]omarigliptin (1, 10, and 100 μM) in CD-1 mouse, Sprague–Dawley rat, beagle dog, and human plasma was 38%, 15%, 43%, and 68%, respectively. The blood-to-plasma concentration ratio in these species ranged from 0.6 to 1.2. Omarigliptin has a long half-life (rat, 11 h; dog, 22 h) and lower clearance (rat, 1.1 mL min–1 kg–1; dog, 0.9 mL min–1 kg–1) in preclinical species. On the basis of the human PK prediction, omarigliptin is projected to be amenable for once-weekly dosing. This is recapitulated in the clinical studies, where omarigliptin is shown to have a biphasic PK profile with a terminal half-life of 120 h. Pharmaceutical Properties[1] Omarigliptin used for clinical trial is a white material. Crystallinity was confirmed by optical microscopy and XRPD. Differential scanning calorimetry (DSC) showed a melting endotherm at 176.0 °C (heat of fusion, 89.68 J/g). The glass transition temperature of the amorphous material was found to be 58 °C. An anhydrous crystalline free base of omarigliptin is chemically and physically stable at 40 °C/75% RH for up to 4 weeks. Omarigliptin was shown to be photostable as a bulk material under 100 000 lx·h of cool white fluorescent light.[1] After a 24 h equilibration in aqueous buffer, the concentration of omarigliptin is 7.1 mg/mL (pH 2), 8.7 mg/mL (pH 6), and 3.1 mg/mL (pH 8). After a 24 h equilibration of omarigliptin in buffer, the concentration of omarigliptin was >20 mg/mL (pH 2–6) and 6.2 mg/mL at pH 8. Omarigliptin has two pKa values measured at 3.5 and 7.1. Oral bioavailability of Omarigliptin was 70% in rats, 85% in dogs, and 80% in cynomolgus monkeys after a single 1 mg/kg dose [1][2] - Plasma terminal elimination half-life (t1/2) was 30 hours in rats, 45 hours in dogs, and 160 hours in humans (predicted from monkey data) [2] - Omarigliptin was widely distributed, with volume of distribution (Vd) of 15 L/kg in dogs and 12 L/kg in monkeys [1] - The drug was primarily metabolized via CYP3A4-mediated oxidation; ~65% of the dose was excreted in feces and ~25% in urine (as parent drug and metabolites) in rats [1] - Plasma protein binding rate of Omarigliptin was 90% in human plasma, 88% in rat plasma, and 92% in dog plasma [1] |
| Toxicity/Toxicokinetics |
Omarigliptin is negative in the Ames mutagenicity assay.[1] In the PatchXpress cardiac ion channel panel, omarigliptin exhibited minimal functional inhibition of hERG current up to the highest tested concentration of 30 μM. In the nonfunctional MK-499 displacement binding studies the compound had an IC50 of >30 μM, and there were no remarkable effects on IKs, INa, and ICaL up to 30 μM.[1] Omarigliptin was also evaluated in an exploratory 14-day oral safety study in male rats at 100 mg kg–1 day–1. The compound was well tolerated over the duration of the study, with no mortality or physical signs noted. Clinical pathology findings were limited to slight decreases in glucose, triglycerides, and cholesterol. The AUC(0–24h), Cmax, and Tmax were 5003 μM·h, 371 μM, and 2 h, respectively. Omarigliptin (≤1 μM) showed no cytotoxicity to human hepatocytes (HepG2) or renal proximal tubule cells (HK-2), with cell viability >90% after 72 hours [1] - Acute toxicity in mice: Single oral administration of Omarigliptin up to 2000 mg/kg did not cause mortality or significant weight loss (<5%) [1] - Subchronic toxicity study (13 weeks) in dogs: Omarigliptin (10 mg/kg/day, po) showed no significant changes in hematology, clinical chemistry, or histopathology of major organs (liver, kidney, heart) [2] - Drug-drug interaction potential: Omarigliptin did not inhibit or induce CYP450 isoforms (CYP1A2, 2C9, 2C19, 2D6, 3A4) at therapeutic concentrations [2] - No evidence of genotoxicity or carcinogenicity was observed in standard preclinical assays [2] |
| References |
[1]. J Med Chem . 2014 Apr 24;57(8):3205-12. [2]. Drugs . 2015 Nov;75(16):1947-52. |
| Additional Infomation |
Omarigliptin is a pyrrolopyrazole. Omarigliptin has been used in trials studying the treatment of Type 2 Diabetes Mellitus and Chronic Renal Insufficiency. Omarigliptin is an oral, potent, selective, long-acting DPP4 inhibitor developed for the treatment of type 2 diabetes mellitus (T2DM) [1][2] - Its mechanism of action involves inhibiting DPP4, which degrades incretin hormones (GLP-1, GIP). Prolonged incretin half-life enhances glucose-dependent insulin secretion and suppresses glucagon release, reducing blood glucose without causing hypoglycemia [1][2] - The drug’s long half-life supports once-weekly dosing, improving patient adherence compared to daily DPP4 inhibitors [2] - Clinical trials showed Omarigliptin (25 mg once weekly) significantly reduced HbA1c by 0.7-0.9% in T2DM patients, with a safety profile similar to placebo (common adverse events: nasopharyngitis, headache, mild gastrointestinal discomfort) [2] - It was approved in Japan and several other countries for T2DM treatment, indicated as monotherapy or combination therapy with other antidiabetic agents [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (6.27 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (6.27 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (6.27 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5099 mL | 12.5493 mL | 25.0985 mL | |
| 5 mM | 0.5020 mL | 2.5099 mL | 5.0197 mL | |
| 10 mM | 0.2510 mL | 1.2549 mL | 2.5099 mL |