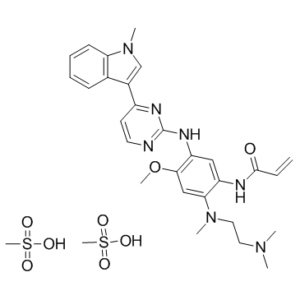
Osimertinib dimesylate (mereletinib; AZD 9291; AZD9291; AZD-9291; trade name Tagrisso), the dimesylate salt of osimertinib, is an oral bioavailable and covalent/irreversible EGFR inhibitor approved as a cancer treatment in 2017 by both the Food and Drug Administration and the European Commission. It inhibits EGFR mutants (WT EGFR, L858R/T790M EGFR, and Exon 19 deletion EGFR) with IC50 values in LoVo cells of 12.92, 11.44, and 493.8 nM, respectively.
Physicochemical Properties
| Molecular Formula | C30H41N7O8S2 |
| Molecular Weight | 691.818644285202 |
| Exact Mass | 691.245 |
| CAS # | 2070014-82-1 |
| Related CAS # | Osimertinib;1421373-65-0;Osimertinib mesylate;1421373-66-1 |
| PubChem CID | 92044416 |
| Appearance | Light yellow to yellow solid |
| Hydrogen Bond Donor Count | 4 |
| Hydrogen Bond Acceptor Count | 13 |
| Rotatable Bond Count | 10 |
| Heavy Atom Count | 47 |
| Complexity | 845 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | RPUCCTLBBCSFEX-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C28H33N7O2.2CH4O3S/c1-7-27(36)30-22-16-23(26(37-6)17-25(22)34(4)15-14-33(2)3)32-28-29-13-12-21(31-28)20-18-35(5)24-11-9-8-10-19(20)24;2*1-5(2,3)4/h7-13,16-18H,1,14-15H2,2-6H3,(H,30,36) |
| Chemical Name | N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide;methanesulfonic acid |
| Synonyms | AZD-9291 dimesylate; AZD9291; AZD 9291; Mereletinib dimesylate; Trade name: Tagrisso |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
EGFRL858R (IC50 = 12 nM); EGFRL858R/T790M (IC50 = 1 nM) Osimertinib (AZD-9291) has mean IC50 values ranging from 13 to 54 nM. It is comparable in potency to early generation tyrosine kinase inhibitors (TKIs) in inhibiting EGFR phosphorylation in EGFR cells harboring sensitizing EGFR mutants such as PC-9 (ex19del), H3255 (L858R), and H1650 (ex19del). With mean IC50 potency less than 15 nM, osimertinib (AZD-9291) also potently inhibits EGFR phosphorylation in T790M mutant cell lines (H1975 (L858R/T790M), PC-9VanR (ex19del/T790M)[1]. |
| ln Vitro |
Osimertinib (AZD-9291) has mean IC50 values ranging from 13 to 54 nM. It is comparable in potency to early generation tyrosine kinase inhibitors (TKIs) in inhibiting EGFR phosphorylation in EGFR cells harboring sensitizing EGFR mutants such as PC-9 (ex19del), H3255 (L858R), and H1650 (ex19del). With mean IC50 potency less than 15 nM, osimertinib (AZD-9291) also potently inhibits EGFR phosphorylation in T790M mutant cell lines (H1975 (L858R/T790M), PC-9VanR (ex19del/T790M)[1]. AZD9291 potently inhibits phosphorylation of EGFR in cell lines harboring sensitizing mutations (PC-9 (ex19del), H3255 (L858R), H1650 (ex19del)) with mean IC50 values ranging from 13 to 54 nM. AZD9291 also potently inhibits phosphorylation of EGFR in T790M mutant cell lines (H1975 (L858R/T790M), PC-9VanR (ex19del/T790M)) with mean IC50 potency less than 15 nM, in contrast to first-generation reversible TKIs which were ineffective. In wild-type EGFR cell lines (A431, LOVO, NCI-H2073), AZD9291 showed less potency with mean IC50 range of 480 to 1865 nM, demonstrating a selectivity margin over wild-type EGFR. Immunoblot analysis confirmed that AZD9291 more potently inhibited phospho-EGFR and downstream signaling substrates (pAKT, pERK) in mutant EGFR cells compared to wild-type. In cell proliferation (Sytox) assays, AZD9291 showed high potency in sensitizing-mutant (mean IC50 of 8 nM in PC-9) and T790M mutant (mean IC50 of 11 nM in H1975, 40 nM in PC-9VanR) EGFR cell lines, with much less activity in wild-type EGFR cell lines (mean IC50 of 650 nM in Calu3, 461 nM in H2073). AZD9291 displayed low nanomolar activity against isogenic cell line pairs harboring EGFR T790M, but was not effective against lines with non-T790M resistance mechanisms (e.g., NRAS mutation, MET amplification, EMT). Chronic treatment of PC-9 cells with AZD9291 did not lead to acquired resistance via T790M mutation, unlike gefitinib or afatinib. AZD9291 showed activity against rare EGFR mutants (G719S, L861Q, an exon 19 insertion) and moderate potency against HER2 mutants (e.g., G776insVC, YVMA ins). It was not effective against EGFR exon 20 insertion mutations. The circulating metabolites AZ5104 and AZ7550 also showed activity, with AZ5104 being more potent against both mutant and wild-type EGFR, thus having a reduced selectivity margin compared to the parent compound AZD9291. [1] |
| ln Vivo |
Osimertinib (AZD-9291) (5 mg/kg/day) is administered to the tumor-bearing mice for a duration of one to two weeks. After receiving Osimertinib (AZD-9291) therapy, 5 out of 5 C/L858R mice showed an almost 80% reduction in tumor volume by magnetic resonance imaging (MRI) within days of treatment, whereas 5 out of 5 mice given a vehicle treatment showed tumor growth[1]. Osimertinib (AZD-9291) is chosen for additional research because it exhibits better rat PK, decreased hERG affinity, and improved IGF1R margins in comparison to the previously reported compounds. Comparing osimertinib (AZD-9291) to the previously reported lead compounds, it also provides a further level of wider chemical and profile diversity. In three efficacy models, Osimertinib (AZD-9291) doses comparatively well; comparable efficacy is seen at doses of 10 mg/kg per day. When Osimertinib (AZD-9291) is dosed at 5 mg/kg per day, even greater efficacy is seen[2]. Once daily oral dosing of AZD9291 induced significant dose-dependent tumor regression in PC-9 (ex19del) and H1975 (L858R/T790M) subcutaneous xenograft models, with shrinkage observed at doses as low as 2.5 mg/kg/day after 14 days. Chronic daily oral dosing (5 mg/kg/day) resulted in complete and durable macroscopic responses in PC-9 and H1975 xenografts, with responses sustained out to 200 days without tumor progression. In a transgenic mouse model with lung adenocarcinomas driven by EGFR L858R (C/L858R), both AZD9291 (5 mg/kg/day) and afatinib induced ~80% reduction in tumor volume. In a transgenic mouse model with tumors driven by EGFR L858R+T790M (C/L+T), only AZD9291 treatment induced significant tumor shrinkage, while afatinib was ineffective. A single dose of AZD9291 (5 mg/kg) in H1975 xenografts strongly inhibited phospho-EGFR and downstream signaling pathways (pERK, pS6, pPRAS40) for up to 30 hours, consistent with its irreversible mechanism. Similar target inhibition was observed in the L858R/T790M transgenic model after AZD9291 dosing via immunohistochemistry and immunoblotting. In A431 (wild-type EGFR amplified) xenografts, AZD9291 (5 mg/kg/day) induced only moderate tumor growth inhibition, in contrast to the profound shrinkage seen in mutant EGFR models at the same dose, confirming its selectivity in vivo. [1] |
| Enzyme Assay |
Osimertinib, formerly known as mereletinib and AZD-9291, is a third generation EGFR inhibitor that is available orally and is irreversible. It selectively targets specific mutants of EGFR, with IC50 values of 493.8 nM for wild-type EGFR in LoVo cells, L858R/T790M EGFR, and Exon 19 deletion EGFR, respectively. It reduces the side effects associated with currently available medications by inhibiting both activating and resistant EGFR mutations while protecting the normal form of EGFR found in normal skin and gut cells. EGFR recombinant enzyme assays were performed to determine inhibitory activity. The amount of active enzyme changes over time, so IC50 values reported are time-dependent apparent IC50s. The assay measured the inhibition of kinase activity of different EGFR constructs (wild-type, L858R, L858R/T790M). [1] |
| Cell Assay |
PC-9 cells are cultured at 37°C with 5% CO2 after being seeded into T75 flasks (5×105 cells/flask) with RPMI growth media. The medium is changed the next day to one supplemented with an EGFR inhibitor at a concentration equivalent to the predetermined EC50 in PC-9 cells. Every two to three days, the medium is changed, and resistant clones are allowed to reach 80% confluency before the cells are trypsinized and reseeded in the same amount of media that contains twice as much EGFR inhibitor. Until a final concentration of 1.5 μM ZD1839, 1.5 μM BIBW 2992, 1.5 μM WZ4002, or 160 nM Osimertinib (AZD-9291) is reached, dose escalations are carried out[1]. For in vitro EGFR phosphorylation assays, cells were treated with a dose-response of each drug for 2 hours. Wild-type cells were stimulated with EGF for 10 minutes before lysis. The level of EGFR phosphorylation was quantified in cell extracts using a modified phospho-EGFR ELISA. For in vitro cell phenotype assays (Sytox proliferation assays), cells were treated with compounds and cell viability/death was assessed. IC50 values were interpolated from the dose-response curves. Growth inhibition assays were performed using a cell viability reagent. Cells were treated with compounds and viability was measured. IC50 values were calculated from the resulting data. For immunoblotting, cells were lysed, proteins were separated by SDS-PAGE, transferred to membranes, and probed with specific antibodies against total and phosphorylated forms of EGFR, AKT, ERK, and other proteins. Signals were detected using chemiluminescence. To generate EGFR inhibitor resistant cell populations, PC-9 cells were continuously exposed to increasing concentrations of the inhibitor (gefitinib, afatinib, WZ4002, or AZD9291) until resistant clones emerged. [1] |
| Animal Protocol |
Mice: The mice used are male and female EGFRL858R and EGFRL858R+T790M mice. Oral gavage is used to administer osimertinib (AZD-9291) at a dose of 7.5 mg/kg and 5 mg/kg, respectively. The drug is suspended in 1% Polysorbate 80. Every week, the Vanderbilt University Institute of Imaging Science images mice. Prior to lung dissection and flash freezing, mice are given a drug treatment for eight hours in preparation for immunoblot analysis. Liquid nitrogen is used to grind the lungs before lysis. Rats: The 10-week-old male RccHan:WIST rats are given a single oral dose of 200 mg/kg of osimertinib (AZD-9291). Accuchek Active meters are used to measure blood glucose levels. For subcutaneous xenograft studies, tumor cells (e.g., PC-9, H1975, A431) were implanted into immunodeficient mice. When tumors reached a specified volume, mice were randomized into treatment groups. Mice were dosed once daily by oral gavage with AZD9291 (suspended in a suitable vehicle) or vehicle control for the duration of the treatment period. Tumor volumes were measured regularly. For pharmacodynamic studies, mice bearing H1975 xenografts were given a single oral dose of AZD9291 (5 mg/kg) or vehicle. Tumors were excised at specified timepoints (1, 6, 16, 24, 30 hours) and processed for immunohistochemical analysis of phospho-proteins. For transgenic mouse studies, bitransgenic mice with tetracycline-inducible expression of EGFR L858R or EGFR L858R+T790M in lung epithelium were used. After tumor development induced by doxycycline, mice were treated via oral gavage once daily with AZD9291 (5 mg/kg), afatinib (7.5 mg/kg), or vehicle control. Tumor response in transgenic mice was assessed weekly by magnetic resonance imaging (MRI) to measure lung tumor volume. For immunoblot analysis of transgenic mouse tumors, mice were treated for eight hours before lungs were harvested, pulverized, and lysed for protein analysis. [1] |
| ADME/Pharmacokinetics |
AZD9291 demonstrated good bioavailability and was widely distributed in tissues after oral dosing in mice. In mice, AZD9291 had moderate clearance with a half-life of around 3 hours. Two active circulating metabolites were identified in vivo: AZ5104 and AZ7550. In mice, the metabolites had similar half-lives to the parent compound. The total exposure (AUC) for AZ7550 and AZ5104 was approximately 68% and 33% of the parent compound, respectively. In a preliminary clinical pharmacokinetic analysis from a Phase I trial, AZD9291, AZ5104, and AZ7550 had a half-life of at least 50 hours in patients, longer than predicted from preclinical data, resulting in a flat PK profile after multiple once-daily doses. Quantitative whole-body autoradiography studies in rats indicated AZD9291 had a brain-to-blood ratio of up to 2 over the first 24 hours, suggesting potential for brain penetration. [1] |
| Toxicity/Toxicokinetics |
In long-term mouse xenograft studies, daily dosing of AZD9291 was well tolerated with minimal body weight loss (less than 5% of starting weight) even after 200 days of treatment. In the two clinical proof-of-principle patients treated with AZD9291 (20 mg once daily), there were no rash events, and only one reported CTCAE Grade 1 diarrhea, consistent with low activity against wild-type EGFR. No significant aberration of blood glucose levels was noted in the two clinical patients, consistent with lack of significant IGF-1R activity. A single oral dose of 200 mg/kg of AZD9291 in rats did not cause a significant change in blood glucose or insulin levels over 24 hours. [1] |
| References |
[1]. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014 Sep;4(9):1046-61. [2]. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistancemutations that spares the wild type form of the receptor. J Med Chem. 2014 Oct 23;57(20):8249-67. |
| Additional Infomation |
The first generation EGFR TKIs gefitinib and erlotinib provide significant clinical benefit in patients with advanced lung adenocarcinoma harbouring activating EGFR mutants (EGFRm+), but patients will ultimately develop disease progression due to acquired resistance. Acquisition of the EGFR T790M mutation is the most common mechanism of drug resistance, detected in more than 50% of gefitinib/erlotinib resistant patients. Current therapeutic strategies are limited for advanced lung adenocarcinoma patients with EGFR T790M (EGFRm+/T790M), so this remains a key area of unmet need. AZD9291 (structure to be disclosed at meeting) is an oral, irreversible, third generation, selective inhibitor of both EGFR activating (EGFRm+) and resistance (EGFRm+/T790M) mutations. The mechanistic and functional activity of AZD9291 was characterised in vitro and in vivo across a number of cell lines harbouring various EGFR-mutations or wild type EGFR. Presented data shows AZD9291 potently inhibits EGFR phosphorylation in EGFRm+ (e.g. PC9; <25nM) and EGFRm+/T790M (e.g. H1975; <25nM) cell lines in vitro, whilst demonstrating much less activity against wild-type EGFR lines (e.g. LoVo; >500nM). Consistently, AZD9291 showed significantly more potent inhibition of proliferation in mutant EGFR cell lines compared to wild-type in vitro. In addition, AZD9291 administered once daily orally at 5mg/kg caused profound regression of tumours across EGFRm+ (PC9; 178% growth inhibition) and EGFRm+/T790M (H1975; 119% growth inhibition) tumour models in vivo, after 14 days dosing. Furthermore 5mg/kg AZD9291 was sufficient to cause significant shrinkage of EGFRm+ and EGFRm+/T790M transgenic mouse lung tumours. Tumour growth inhibition was associated with profound inhibition of EGFR phosphorylation and key downstream signaling pathways such as AKT and ERK. Chronic long-term treatment of PC9 and H1975 xenograft tumours with AZD9291 led to a complete and sustained macroscopic response, with no visible tumours after 40 days dosing, and being maintained beyond 100 days. Furthermore, pre-clinical data also indicates that AZD9291 could target tumours that have acquired resistance to the more recently identified HER2-amplification mechanism, thus potentially extending its benefit in TKI resistant patients. Taken together, preclinical data demonstrates that AZD9291 is a potent and effective inhibitor of both EGFR activating (EGFRm+) and resistance (EGFRm+/T790M) mutations whilst sparing wild-type EGFR. These data support the further clinical investigation of AZD9291 in advanced EGFR mutant lung adenocarcinoma. AZD9291 (osimertinib) is a novel, oral, potent, irreversible third-generation EGFR tyrosine kinase inhibitor. It is structurally distinct from other third-generation TKIs (WZ4002, CO-1686), being a mono-anilino-pyrimidine compound. It covalently binds to the cysteine-797 residue in the ATP-binding site of EGFR, leading to irreversible inhibition. It was designed to selectively inhibit both EGFR sensitizing mutations (ex19del, L858R) and the T790M resistance mutation while sparing wild-type EGFR, aiming to reduce side effects like rash and diarrhea. Proof-of-principle clinical activity was shown in two advanced NSCLC patients with EGFR T790M-mediated resistance to prior EGFR TKIs. Both patients achieved confirmed radiographic partial responses at the lowest dose (20 mg once daily) with a mild side effect profile. Preclinical data suggests AZD9291 may have potential to treat brain metastases due to favorable brain penetration in rat models. Resistance to AZD9291 in vitro did not occur via T790M acquisition, suggesting future resistance may involve non-EGFR pathways (e.g., HER2 amplification, MET amplification, phenotypic transformation). [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
|
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.4455 mL | 7.2273 mL | 14.4546 mL | |
| 5 mM | 0.2891 mL | 1.4455 mL | 2.8909 mL | |
| 10 mM | 0.1445 mL | 0.7227 mL | 1.4455 mL |