Physicochemical Properties
| Molecular Formula | C14H17N3 |
| Molecular Weight | 227.1422 |
| Exact Mass | 459.164 |
| Elemental Analysis | C, 73.98; H, 7.54; N, 18.49 |
| CAS # | 28614-26-8 |
| Related CAS # | 171205-17-7 |
| PubChem CID | 5013 |
| Appearance | Typically exists as solid at room temperature |
| LogP | 1.413 |
| Hydrogen Bond Donor Count | 0 |
| Hydrogen Bond Acceptor Count | 3 |
| Rotatable Bond Count | 1 |
| Heavy Atom Count | 17 |
| Complexity | 248 |
| Defined Atom Stereocenter Count | 0 |
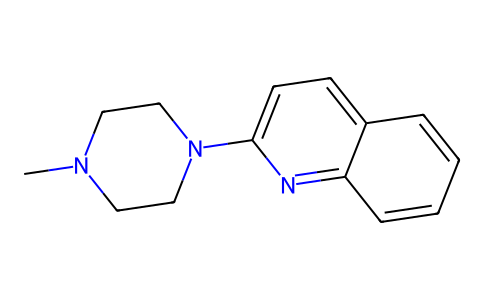
| SMILES | CN1CCN(C2C=CC3C(=CC=CC=3)N=2)CC1 |
| InChi Key | HOMWNUXPSJQSSU-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C14H17N3/c1-16-8-10-17(11-9-16)14-7-6-12-4-2-3-5-13(12)15-14/h2-7H,8-11H2,1H3 |
| Chemical Name | 2-(4-methylpiperazin-1-yl)quinoline |
| Synonyms | N Methylquipazine; NMethylquipazine; 2-(4-methylpiperazin-1-yl)quinoline; 28614-26-8; quinoline, 2-(4-methyl-1-piperazinyl)-; 0YV1ZIR6S0; CHEMBL288591; CHEBI:64164; 1-(2-Quinolyl)-4-methylpiperazine; N-Methylquipazine |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | 5-HT3 receptor |
| ln Vitro | N-Methylquipazine (2-[1-(4-methyl)-piperazinyl)quinoline)) was labelled with carbon-11 by reacting [11C]methyl iodide with the nor-compound, quipazine. Radiochemical conversions were 79 +/- 7%, based on the alkylating agent. The total synthesis time including purification was 40 to 45 min. N-[Methyl-11C]methylquipazine thus synthesized was >99% radiochemically pure, and the specific activity ranged between 12-37 GBq/mumol [2]. |
| ln Vivo |
In this study, researchers examined the effect of n-methylquipazine (NMQ), which is a putative 5-hydroxytryptamine3(5-HT3)receptor agonist, on the extracellular concentrations of dopamine (DA) and one of its metabolites, dihydroxyphenylacetic acid (DOPAC), in the anterior medial prefrontal cortex (AmPFc) of awake, freely moving rats. The administration of NMQ via the perfusion fluid produced a concentration-dependent (10-1,000 microM) increase in extracellular DA levels in the AmPFc. In contrast, NMQ produced a decrease in the extracellular concentrations of DOPAC. The increase in extracellular DA levels returned to baseline after the removal of NMQ from the perfusate. The increase in extracellular DA levels in the AmPFc produced by 100 microM of NMQ was markedly attenuated by either the coadministration of tetrodotoxin (1 microM), which inhibits axonal impulse flow, or the depletion of extracellular Ca2+ by removing CaCl2 and adding EDTA to the perfusate. The intradialysate administration of the 5-HT3 antagonist BRL 46470A produced a concentration-dependent (10-1,000 microM) decrease in extracellular DA levels, and this effect was reversible on removal from the perfusate. In contrast, ondansetron (500 and 1,000 microM), which is another 5-HT3 receptor antagonist, produced a transient increase followed by a sustained decrease in extracellular DA levels. The preinfusion of 10 microM of BRL 46470 followed by coperfusion of BRL 46470A with 50 or 100 microM of NMQ via the dialysis probe did not significantly attenuate the increase of NMQ in extracellular DA levels in the AmPFc. The administration of the selective 5-HT2 receptor MDL 100907 (1 mg/kg, i.p.) also did not alter the increase in basal DA levels produced by 100 microM of NMQ. The pretreatment of rats with alpha-methyl-p-tyrosine produced a significant attenuation in the NMQ-induced increase in extracellular DA levels, suggesting that the elevation by NMQ of DA levels is dependent on newly synthesized stores of DA. Overall, these results suggest that the increase in AmPFc DA levels by NMQ is probably not mediated by its interaction with the 5-HT3 receptor [1]. Dynamic imaging with PET was used to examine in vivo its distribution in rat and monkey. In rat the organ uptake at intermediate times was: liver > heart > whole brain > or = lung > extracerebral tissue. Brain uptake and wash-out were rapid: A maximum was reached in 2 to 3 min with subsequent decrease to approximately equal to 50% the peak value by 13 min. In monkey the tracer uptake was heterogeneous and high in regions known to contain 5-HT3 receptors but also in regions devoid of these receptors. Tissue kinetics were similar for all regions (initial rapid accumulation with tmax < or = 7 min, followed by slow decrease with all regions approaching the level of the cerebellum at 30 to 35 min). Pretreating with quipazine significantly decreased only the ratio of uptake in the medulla oblongata compared to the cerebellum. Although the nonspecificity of its binding limits the usefulness of N-[methyl-11C]methylquipazine, both its kinetic behavior and the blocking results indicate that a more selective arylpiperazine might prove to be a more attractive tracer for PET studies of 5-HT3 receptors[2]. |
| References |
[1]. The characterization of the effect of locally applied N-methylquipazine, a 5-HT3 receptor agonist, on extracellular dopamine levels in the anterior medial prefrontal cortex in the rat: an in vivo microdialysis study. Synapse. 1996 Dec;24(4):313-21. [2]. N-methylquipazine: carbon-11 labelling of the 5-HT3 agonist and in vivo evaluation of its biodistribution using PETNucl Med Biol. 1997 Jul;24(5):405-12. |
| Additional Infomation | N-methylquipazine is an aminoquinoline that consists of quinoline in which the hydrogen at position 2 is substituted by a 4-methylpiperazin-1-yl group. A 5-HT3 agonist. Has almost the same affinity for 5-HT3 sites as quipazine but unlike the latter, does not bind to 5-HT1B sites. It has a role as a serotonergic agonist. It is a N-alkylpiperazine, a N-arylpiperazine and an aminoquinoline. |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 4.4026 mL | 22.0129 mL | 44.0257 mL | |
| 5 mM | 0.8805 mL | 4.4026 mL | 8.8051 mL | |
| 10 mM | 0.4403 mL | 2.2013 mL | 4.4026 mL |