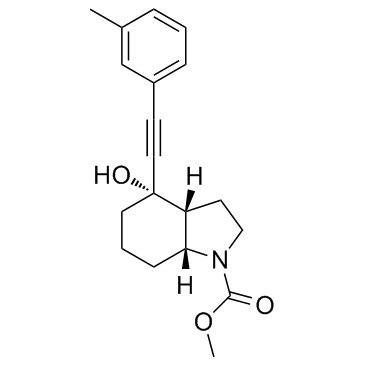
Mavoglurant (AFQ-056; AFQ056) is a potent and non-competitive mGlu5 (metabotropic glutamate receptor 5) receptor antagonist (IC50=30 nM) with the potential to be used for treatment of fragile X syndrome. It showed efficacy in the treatment of L-dopa induced dyskinesias in Parkinson's disease and Fragile X mental retardation in proof of principle studies.
Physicochemical Properties
| Molecular Formula | C19H23NO3 |
| Molecular Weight | 313.397 |
| Exact Mass | 313.167 |
| CAS # | 543906-09-8 |
| Related CAS # | Mavoglurant racemate;1636881-61-2 |
| PubChem CID | 9926832 |
| Appearance | White to off-white solid powder |
| Density | 1.2±0.1 g/cm3 |
| Boiling Point | 476.3±45.0 °C at 760 mmHg |
| Flash Point | 241.8±28.7 °C |
| Vapour Pressure | 0.0±1.3 mmHg at 25°C |
| Index of Refraction | 1.602 |
| LogP | 3.51 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 3 |
| Rotatable Bond Count | 3 |
| Heavy Atom Count | 23 |
| Complexity | 519 |
| Defined Atom Stereocenter Count | 3 |
| SMILES | CC1=CC(=CC=C1)C#C[C@@]2(CCC[C@@H]3[C@H]2CCN3C(=O)OC)O |
| InChi Key | ZFPZEYHRWGMJCV-ZHALLVOQSA-N |
| InChi Code | InChI=1S/C19H23NO3/c1-14-5-3-6-15(13-14)8-11-19(22)10-4-7-17-16(19)9-12-20(17)18(21)23-2/h3,5-6,13,16-17,22H,4,7,9-10,12H2,1-2H3/t16-,17-,19-/m1/s1 |
| Chemical Name | methyl (3aR,4S,7aR)-4-hydroxy-4-[2-(3-methylphenyl)ethynyl]-3,3a,5,6,7,7a-hexahydro-2H-indole-1-carboxylate |
| Synonyms | AFQ 056; AFQ-056; AFQ056 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
- Mavoglurant (AFQ056) selectively targets metabotropic glutamate receptor 5 (mGluR5) as a negative allosteric modulator; the Ki value was reported as 14 nM in radioligand binding assays [1] - No significant binding affinity was observed for other mGluR subtypes (mGluR1/2/3/4/6/7/8) at concentrations up to 10 μM [1] |
| ln Vitro |
- In HEK293 cells expressing human mGluR5, Mavoglurant (AFQ056) inhibited quisqualate-induced calcium mobilization with an IC50 of 33 nM [1] - It dose-dependently reduced glutamate-induced inositol phosphate accumulation in rat cortical neurons, with maximal inhibition (72%) at 1 μM [1] - In primary fibroblasts from fragile X syndrome patients, Mavoglurant (AFQ056) (1 μM) normalized excessive protein synthesis caused by FMR1 gene silencing [2] Mavoglurant (1 nM-10 μM; 10 min) totally antagonizes hmGluR5-mediated responses with IC50 of 110 and 30 nM, respectively, in Ca2+- and PI turnover tests in L(tk-) cells stably expressing mGluR5a [1] . Mavoglurant (0.01 nM-10 μM) displaces the binding of the allosteric binding ligand [3H]-AAE327 in a concentration-dependent manner in rat meninges with an IC50 of 47 nM [1]. |
| ln Vivo |
- In Fmr1 knockout mice (fragile X syndrome model), oral administration of Mavoglurant (AFQ056) (10 mg/kg) significantly improved audiogenic seizures (70% reduction) and hyperactivity (35% decrease in locomotor activity) [1] - It restored hippocampal long-term depression (LTD) in Fmr1 knockout mice at doses ≥3 mg/kg, as measured by field excitatory postsynaptic potential (fEPSP) recordings [1] - In a rat model of Parkinson's disease induced by 6-OHDA lesion, Mavoglurant (AFQ056) (3 mg/kg, i.p.) reduced levodopa-induced dyskinesia by 45% without affecting antiparkinsonian efficacy [3] Stress-induced hyperthermia (SIH) in mice is inhibited by mavoglurant (0.1–10 mg/kg; single oral dosage) in a dose-dependent manner [1]. Mavoglurant, at a single oral dose of 9.4 mg/kg, has a terminal half-life of 2.9 hours, a moderate oral bioavailability of 32%, and a Cmax of 950 pmol/mL and 3500 pmol/g in the brain and plasma, respectively [1]. Mavoglurant (3.1 mg/kg; intravenous injection; single dose) has a Tmax of less than 0.08 hours, a Cmax (plasma; brain) of 3330 pmol/mL, and a terminal half-life of 0.69 hours [1]. |
| Enzyme Assay |
- Radioligand binding assay:
- Membrane preparations from mGluR5-expressing cells were incubated with [³H]MPEP (a selective mGluR5 ligand) and serial dilutions of Mavoglurant (AFQ056) (0.01-1000 nM) in binding buffer (pH 7.4) for 90 minutes at 25°C.
- Bound ligand was separated by filtration, and radioactivity was measured to calculate displacement potency and Ki value [1] - Calcium mobilization assay: - mGluR5-transfected HEK293 cells loaded with calcium-sensitive dye were stimulated with quisqualate (1 μM) in the presence of Mavoglurant (AFQ056). - Fluorescence intensity was monitored for 300 seconds to determine IC50 for receptor-mediated calcium response inhibition [1] |
| Cell Assay |
- Protein synthesis assay in fragile X fibroblasts:
- Primary fibroblasts from patients with FMR1 full mutation were treated with Mavoglurant (AFQ056) (0.1-10 μM) for 24 hours.
- Protein synthesis was measured by [³H]leucine incorporation, with normalization to total protein content [2] - Inositol phosphate accumulation assay: - Rat cortical neurons were preincubated with Mavoglurant (AFQ056) for 30 minutes, followed by glutamate stimulation (100 μM) for 1 hour. - Accumulated inositol phosphates were extracted and quantified using scintillation counting [1] |
| Animal Protocol |
Animal/Disease Models: Male OF1/IC mice [1] Doses: 0.1, 1, 10 mg/kg Route of Administration: Single oral administration Experimental Results: Reduce stress-induced hyperthermia. It is equivalent to the positive control chlordiazepoxide. Animal/Disease Models: Male SD (SD (Sprague-Dawley)) rat (175-250 g) [1] Doses: 3.1 mg/kg intravenously (iv) (iv)(iv); 9.4 mg/kg orally (pharmacokinetic/PK/PK analysis) Route of Administration: Single iv or oral administration Experimental Results: Po: F=32%; T1/2=2.9 hrs (hrs (hours)); Tmax≤0.25 hrs (hrs (hours)). IV: T1/2=0.69h; Cmax (plasma/brain)=3330 pmol·mL-1/8400 pmol·g-1; Tmax≤0.08 hour. - Fragile X syndrome mouse model: - Male Fmr1 knockout mice (8-12 weeks old) received oral Mavoglurant (AFQ056) suspended in 0.5% methylcellulose at doses of 1, 3, or 10 mg/kg once daily for 7 days. - Behavioral tests (open field, audiogenic seizure susceptibility) were conducted 1 hour after the final dose, followed by hippocampal tissue collection for electrophysiological analysis [1] - Parkinson's disease rat model: - Rats with unilateral 6-OHDA lesions received intraperitoneal injections of Mavoglurant (AFQ056) (1 or 3 mg/kg) 30 minutes prior to levodopa (6 mg/kg) administration, daily for 21 days. - Dyskinesia severity was scored using abnormal involuntary movement (AIM) scales during the treatment period [3] |
| ADME/Pharmacokinetics |
- After oral administration of Mavoglurant (AFQ056) (10 mg/kg) in mice, peak plasma concentration (Cmax) was reached at 1.2 hours with a bioavailability of 65% [1] - The compound showed good brain penetration with a brain-to-plasma ratio of 2.3 in rats [1] - Plasma elimination half-life was approximately 4.5 hours in rodents [1] |
| Toxicity/Toxicokinetics |
- In 28-day repeated-dose toxicity studies in rats, Mavoglurant (AFQ056) at doses up to 30 mg/kg/day did not induce significant changes in body weight, hematological parameters, or liver/kidney function markers [1] - No evidence of mutagenicity or genotoxicity was observed in standard in vitro assays [1] - Plasma protein binding was >99% in human and rat plasma [1] |
| References |
[1]. AFQ056/mavoglurant, a novel clinically effective mGluR5 antagonist: identification, SAR and pharmacological characterization. Bioorg Med Chem. 2014 Nov 1;22(21):5790-5803. [2]. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. 2011 Jan 5;3(64):64ra1. [3]. Mavoglurant as a treatment for Parkinson's disease. Expert Opin Investig Drugs. 2014 Aug;23(8):1165-79. |
| Additional Infomation |
- Mavoglurant (AFQ056) was identified through structure-activity relationship (SAR) optimization of pyridine derivatives, with improved metabolic stability compared to earlier mGluR5 antagonists [1] - In fragile X syndrome patients, response to Mavoglurant (AFQ056) correlated with FMR1 gene methylation status, with patients showing <5% methylation exhibiting better behavioral improvement [2] - Clinical development for Parkinson's disease focused on reducing levodopa-induced dyskinesia, with Phase II trials showing significant efficacy at doses of 100-200 mg/day [3] Mavoglurant has been used in trials studying the treatment of Patient Diagnosed With OCD and and Resistant to SSRI Treatment (Failed SSRI Over 12 Weeks at Appropriate Doses). |
Solubility Data
| Solubility (In Vitro) | DMSO : ~120 mg/mL (~382.91 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 3 mg/mL (9.57 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 30.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 3 mg/mL (9.57 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 30.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 3 mg/mL (9.57 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 30.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.1908 mL | 15.9541 mL | 31.9081 mL | |
| 5 mM | 0.6382 mL | 3.1908 mL | 6.3816 mL | |
| 10 mM | 0.3191 mL | 1.5954 mL | 3.1908 mL |