MK-2461 (MK2461) is a novel, potent, ATP-competitive, multi-targeted inhibitor for c-Met (WT/mutants) with potential antineoplastic activity. With an IC50 of 0.4-2.5 nM, it inhibits a range of c-Met mutants and has a lower potency against kinases like Ron and Flt1. When it comes to inhibiting c-Met, MK-2461 is 8–30 times more selective than FGFR1, FGFR2, FGFR3, PDGFRβ, KDR, Flt3, Flt4, TrkA, and TrkB. NK MK-2461 efficiently inhibited autophosphorylation of the c-Met activation loop while suppressing constitutive or ligand-induced phosphorylation of the juxtamembrane domain and COOH-terminal docking site of c-Met, as well as its downstream signaling to the phosphoinositide 3-kinase–AKT and Ras–extracellular signal-regulated kinase pathways in tumor cells.
Physicochemical Properties
| Molecular Formula | C24H25N5O5S | |
| Molecular Weight | 495.55 | |
| Exact Mass | 495.157 | |
| Elemental Analysis | C, 58.17; H, 5.09; N, 14.13; O, 16.14; S, 6.47 | |
| CAS # | 917879-39-1 | |
| Related CAS # |
|
|
| PubChem CID | 44137946 | |
| Appearance | Light yellow to yellow solid powder | |
| Density | 1.5±0.1 g/cm3 | |
| Boiling Point | 732.4±70.0 °C at 760 mmHg | |
| Flash Point | 396.7±35.7 °C | |
| Vapour Pressure | 0.0±2.4 mmHg at 25°C | |
| Index of Refraction | 1.693 | |
| LogP | -0.52 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 9 | |
| Rotatable Bond Count | 6 | |
| Heavy Atom Count | 35 | |
| Complexity | 895 | |
| Defined Atom Stereocenter Count | 1 | |
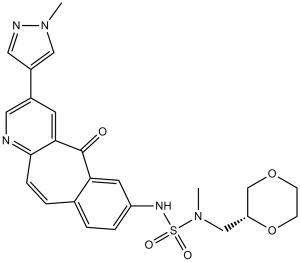
| SMILES | O=C1C2C=C(C=CC=2C=CC2N=CC(C3C=NN(C)C=3)=CC1=2)NS(=O)(=O)N(C)C[C@H]1OCCOC1 |
|
| InChi Key | JGEBLDKNWBUGRZ-HXUWFJFHSA-N | |
| InChi Code | InChI=1S/C24H25N5O5S/c1-28-13-18(12-26-28)17-9-22-23(25-11-17)6-4-16-3-5-19(10-21(16)24(22)30)27-35(31,32)29(2)14-20-15-33-7-8-34-20/h3-6,9-13,20,27H,7-8,14-15H2,1-2H3/t20-/m1/s1 | |
| Chemical Name | 14-[[[(2R)-1,4-dioxan-2-yl]methyl-methylsulfamoyl]amino]-5-(1-methylpyrazol-4-yl)-2-oxo-7-azatricyclo[9.4.0.03,8]pentadeca-1(11),3(8),4,6,9,12,14-heptaene | |
| Synonyms | MK 2461; MK-2461; MK2461 | |
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
c-Met(M1250T) (IC50 = 0.4 nM); c-Met(Y1235D) (IC50 = 0.5 nM); c-Met(Y1230H) (IC50 = 1.0 nM); c-Met(N1100) (IC50 = 1.5 nM); c-Met(Y1230C) (IC50 = 1.5 nM) MK-2461 is a multitargeted kinase inhibitor that preferentially inhibits the activated c-Met receptor (IC₅₀ = 0.4 nM). It also inhibits RON kinase (IC₅₀ = 1.6 nM), VEGFR2 (IC₅₀ = 12 nM), PDGFRβ (IC₅₀ = 25 nM), and c-Kit (IC₅₀ = 38 nM), with no significant activity against EGFR or Abl (IC₅₀ > 1000 nM) [1] |
| ln Vitro |
MK-2461 additionally has IC50 values of 65 nM, 39 nM, 50 nM, 44 nM, 46 nM, 61 nM, and 78 nM for FGFR1, FGFR2, FGFR3, KDR, TrkA, TrkB, and Flt4, respectively. With IC50 values of 1.5 nM, 1.5 nM, 1.0 nM, 0.5 nM, and 0.4 nM, respectively, MK-2461 more effectively suppresses the activity of oncogenic c-Met kinase mutants, including N1100Y, Y1230C, Y1230H, Y1235D, and M1250T, in comparison to wild-type c-Met. Phosphorylated c-Met is more strongly bound by MK-2461 than unphosphorylated c-Met. MK-2461 effectively prevents ATP from causing the c-Met'sCOOH-terminaldocking domain to become autophosphorylated, but not the activation loop. On the other hand, MK-2461 has an IC50 of less than 0.3 μM and prevents phosphorylation of PDGFRα (Y849) in H1703 cells and the activation loop of FGFR2 (Y653/Y654) in Kato III cells. MK-2461 inhibits HGF-induced branching tubulogenesis of MDCK cells, HGF-induced migration of HPAF II cells (IC50 of 404 nM), and HGF-induced mitogenesis of 4MBr-5 cells (IC50 of 204 nM). Furthermore, with an IC50 of approximately 100 nM, MK-2461 effectively suppresses the IL-3-independent proliferation of 32D cells transformed with Tpr-Met or Tpr-Met (Y362C) mutant. A wide panel of tumor cell lines is considerably inhibited in growth by MK-2461, which is particularly effective against tumor cells that have MET or FGFR2 genomic amplification.[1] MK-2461 dose-dependently inhibited proliferation of c-Met-overexpressing tumor cell lines: U-87 MG/c-Met (glioblastoma, IC₅₀ = 0.015 μM), H441 (lung adenocarcinoma, IC₅₀ = 0.02 μM), and MKN-45 (gastric cancer, IC₅₀ = 0.025 μM). It had minimal effect on c-Met-low cells (e.g., parental U-87 MG, IC₅₀ = 5.8 μM) [1] At concentrations ≥ 0.05 μM, the drug potently blocked hepatocyte growth factor (HGF)-induced c-Met phosphorylation (Tyr1234/1235) and downstream signaling (AKT Ser473, ERK1/2 Thr202/Tyr204) in U-87 MG/c-Met cells, as confirmed by Western blot [1] It induced G1 phase cell cycle arrest and apoptosis in H441 cells with an EC₅₀ of 0.08 μM, upregulating cleaved caspase-3/PARP and downregulating cyclin D1 [1] In MKN-45 cell migration/invasion assays, MK-2461 (0.03 μM) reduced migration by ~70% and invasion by ~65% by inhibiting c-Met-mediated MMP-9 expression [1] |
| ln Vivo |
MK-2461 treatment, with an IC50 of approximately 1 μM, significantly inhibits the phosphorylation of c-Met (Y1349) in GTL-16 tumors. MK-2461 can effectively suppress the growth of GTL-16 xenograft tumors in mice by 62%, 77%, 75%, and 90% when given orally at doses of 10 mg/kg, 50 mg/kg, 100 mg/kg, and 200 mg/kg once daily. The growth of NIH3T3 tumors containing the c-Met single nucleotide mutants T3936C and T3997C is similarly inhibited by MK-2461 treatment at 134 mg/kg twice daily, by 78% and 62%, respectively.[1] MK-2461 significantly inhibited tumor growth in nude mice bearing U-87 MG/c-Met xenografts. Oral administration of 25 mg/kg/day for 21 days reduced tumor volume by ~78% vs. controls, with almost complete suppression of intratumoral c-Met phosphorylation [1] In a murine model of H441 xenografts, the drug (30 mg/kg/day, oral for 28 days) achieved a 72% tumor growth inhibition rate and prolonged median survival by 40% [1] It exhibited excellent tumor penetration: at 4 hours post-oral administration (25 mg/kg), tumor-to-plasma concentration ratio was 3.5, maintaining effective drug levels (>0.05 μM) in tumors for 12 hours [1] |
| Enzyme Assay |
EQEDEPEGDYFEWLE-CONH2 phosphorylation is measured by means of a time-resolved fluorescence resonance energy transfer assay. This phosphorylation is catalyzed by c-Met. In [2] Timing-resolved fluorescence resonance energy transfer assays, akin to the c-Met kinase assay, are used to determine the MK-2461 IC50 for Ron, Mer, Flt1, Flt3, Flt4, KDR, PDGFRβ, FGFR1, FGFR2, FGFR3, TrkA, and TrkB. Recombinant activated c-Met kinase domain (Tyr1234/1235 phosphorylated) was incubated with serial dilutions of MK-2461 (0.0001-1 μM) in kinase buffer containing ATP (10 μM) and a biotinylated peptide substrate (c-Met-specific sequence). The reaction was conducted at 37°C for 60 minutes, and phosphorylated substrate was detected via homogeneous time-resolved fluorescence (HTRF) using anti-phosphotyrosine antibody. Inhibition rates were calculated by comparing fluorescence intensity with vehicle controls, and IC₅₀ values were derived from dose-response curves [1] To assess selectivity, recombinant RON, VEGFR2, PDGFRβ, and c-Kit kinase domains were tested using the same protocol. Reaction conditions (buffer composition, temperature, incubation time) were identical, and IC₅₀ values were determined to confirm preferential targeting of activated c-Met [1] |
| Cell Assay |
For 72 hours, cells are exposed to different MK-2461 concentrations. The ViaLight PLUS kit is used to measure the viability of tumor cells. U-87 MG/c-Met, H441, and MKN-45 cells were seeded in 96-well plates (5×10³ cells/well) and treated with MK-2461 (0.001-10 μM) for 72 hours. Cell viability was measured via tetrazolium-based (MTT) assay, and IC₅₀ values were calculated [1] For Western blot analysis: U-87 MG/c-Met cells were serum-starved for 16 hours, treated with 0.01-0.2 μM MK-2461 for 1 hour, then stimulated with HGF (50 ng/mL) for 10 minutes. Cells were lysed, and lysates were probed with antibodies against p-c-Met (Tyr1234/1235), p-AKT (Ser473), p-ERK1/2 (Thr202/Tyr204), cleaved caspase-3, cleaved PARP, cyclin D1, and GAPDH (loading control) [1] Cell migration/invasion assays used Boyden chambers: MKN-45 cells were treated with 0.01-0.1 μM MK-2461 for 1 hour, seeded in upper chambers (uncoated for migration, Matrigel-coated for invasion), and HGF (20 ng/mL) was added to lower chambers. After 24 hours, migrated/invasive cells were fixed, stained, and counted [1] |
| Animal Protocol |
Female nude CD-1 nu/nu mice inoculated s.c. with GTL-16 cells or c-Met mutant-transformed NIH3T3 cells ~134 mg/kg Orally once or twice daily Nude mice (6-8 weeks old) were subcutaneously implanted with U-87 MG/c-Met cells (5×10⁶ cells/mouse) to establish xenografts. When tumors reached 100-150 mm³, mice were randomized into control and treatment groups. MK-2461 was suspended in 0.5% carboxymethylcellulose (CMC) and administered orally at 25 mg/kg/day for 21 days. Tumor volume was measured every 3 days using calipers (volume = length × width² / 2) [1] For H441 xenograft studies: nude mice were implanted with H441 cells (4×10⁶ cells/mouse), and MK-2461 was given orally at 30 mg/kg/day for 28 days. Survival time was recorded daily, and tumors were harvested at sacrifice for p-c-Met immunohistochemistry [1] For pharmacokinetic sampling: mice received a single oral dose of MK-2461 (25 mg/kg), and blood/plasma samples were collected at 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours post-dose. Tumor samples were collected at 4 hours post-dose to measure drug concentration [1] |
| ADME/Pharmacokinetics |
MK-2461 had an oral bioavailability of ~65% in mice after a single 25 mg/kg dose. Maximum plasma concentration (Cmax) was 4.8 μg/mL achieved at 1 hour post-administration, and plasma half-life (t₁/₂) was ~6.2 hours [1] The drug was widely distributed in tissues, with highest concentrations in liver, kidneys, and tumors. Tumor-to-plasma concentration ratio was 3.5 at 4 hours post-dose [1] It was metabolized primarily by cytochrome P450 3A4 (CYP3A4) in human liver microsomes, with a metabolic clearance rate of 0.9 mL/min/mg protein [1] |
| Toxicity/Toxicokinetics |
Mice treated with MK-2461 at 25 mg/kg/day for 21 days showed mild weight loss (~6%) but no significant liver or kidney toxicity. Serum ALT, AST, creatinine, and BUN levels were within normal ranges [1] Plasma protein binding rate of MK-2461 was ~95% in human plasma, determined via equilibrium dialysis [1] In vitro cytotoxicity assays: the drug showed no significant damage to normal human hepatocytes (LO2) or bronchial epithelial cells (BEAS-2B) at concentrations up to 1 μM [1] |
| References |
[1]. MK-2461, a novel multitargeted kinase inhibitor, preferentially inhibits the activated c-Met receptor. Cancer Res. 2010 Feb 15;70(4):1524-33. |
| Additional Infomation |
MK-2461 is a member of the class of benzocycloheptapyridines that is 5H-benzo[4,5]cyclohepta[1,2-b]pyridin-5-one substituted by 1-methyl-1H-pyrazol-4-yl and [{[(2R)-1,4-dioxan-2-yl]methyl}(methyl)sulfamoyl]amino groups at positions 3 and 7, respectively. It is a novel ATP-competitive multi-targeted inhibitor of activated c-Met (IC50 = 2.5 nM). It has a role as an antineoplastic agent, a c-Met tyrosine kinase inhibitor and an apoptosis inducer. It is a member of pyrazoles, a benzocycloheptapyridine, a member of dioxanes and a member of sulfamides. MK-2461 is a novel multitargeted kinase inhibitor designed to preferentially bind to the activated (phosphorylated) conformation of c-Met, minimizing off-target activity against inactive kinases [1] It was developed to target c-Met-driven cancers, including glioblastoma, lung adenocarcinoma, and gastric cancer—tumors often resistant to conventional therapies due to c-Met overexpression or amplification [1] Preclinical data demonstrate that its selectivity for activated c-Met reduces on-target toxicities compared to non-selective c-Met inhibitors, supporting its potential as a targeted therapy for c-Met-addicted tumors [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.04 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.04 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: 1% DMSO+30% polyethylene glycol+1% Tween 80, pH 4: 15mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0180 mL | 10.0898 mL | 20.1796 mL | |
| 5 mM | 0.4036 mL | 2.0180 mL | 4.0359 mL | |
| 10 mM | 0.2018 mL | 1.0090 mL | 2.0180 mL |