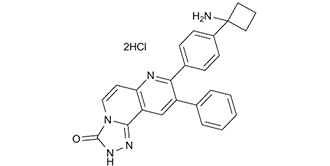
MK-2206 dihydrochloride (2HCl) is a novel, potent, orally bioavailable and highly selective allosteric inhibitor of the serine/threonine protein kinase Akt1/2/3 with potential anticancer activity. In cell-free assays, it inhibits Akt1/2/3 with IC50 values of 8 nM, 12 nM, and 65 nM, respectively; it has little to no inhibitory activity against 250 other protein kinases. By preventing Akt's Thr308 and Ser473 phosphorylation, MK-2206 may have anticancer effects. Akt signaling is suppressed by MK-2206, which also promotes cancer cell death when used alone or in combination with other chemotherapeutic drugs. MK-2206 increases sensitivity to rapamycin by enhancing apoptosis and sensitivity to reactive oxygen species.
Physicochemical Properties
| Molecular Formula | C25H23CL2N5O |
| Molecular Weight | 480.39 |
| Exact Mass | 479.127 |
| Elemental Analysis | C, 62.24; H, 5.22; Cl, 14.70; N, 14.52; O, 3.32 |
| CAS # | 1032350-13-2 |
| Related CAS # | MK-2206 free base;1032349-93-1;MK-2206;1032349-77-1 |
| PubChem CID | 24964624 |
| Appearance | Yellow solid powder |
| LogP | 6.547 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 4 |
| Rotatable Bond Count | 3 |
| Heavy Atom Count | 31 |
| Complexity | 760 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | O=C1N([H])N=C2C3=C([H])C(C4C([H])=C([H])C([H])=C([H])C=4[H])=C(C4C([H])=C([H])C(=C([H])C=4[H])C4(C([H])([H])C([H])([H])C4([H])[H])N([H])[H])N=C3C([H])=C([H])N21 |
| InChi Key | HWUHTJIKQZZBRA-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C25H21N5O.2ClH/c26-25(12-4-13-25)18-9-7-17(8-10-18)22-19(16-5-2-1-3-6-16)15-20-21(27-22)11-14-30-23(20)28-29-24(30)31;;/h1-3,5-11,14-15H,4,12-13,26H2,(H,29,31);2*1H |
| Chemical Name | 8-[4-(1-aminocyclobutyl)phenyl]-9-phenyl-2H-[1,2,4]triazolo[3,4-f][1,6]naphthyridin-3-one;dihydrochloride |
| Synonyms | MK2206 HCl; MK2206 dihydrochloride; MK2206; MK-2206 dihydrochloride; MK-2206 2HCl; MK2206; 8-(4-(1-Aminocyclobutyl)phenyl)-9-phenyl-[1,2,4]triazolo[3,4-f][1,6]naphthyridin-3(2H)-one dihydrochloride; 8-[4-(1-AMINOCYCLOBUTYL)PHENYL]-9-PHENYL-1,2,4-TRIAZOLO[3,4-F][1,6]NAPHTHYRIDIN-3(2H)-ONE DIHYDROCHLORIDE; Q34I3E28IO; 8-[4-(1-aminocyclobutyl)phenyl]-9-phenyl-2H-[1,2,4]triazolo[3,4-f][1,6]naphthyridin-3-one;dihydrochloride; MK 2206; MK-2206 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Akt1 (IC50 = 8 nM); Akt2 (IC50 = 12 nM); Akt3 (IC50 = 65 nM) 1. AKT isoforms (AKT1, AKT2, AKT3) (Literatures [2], [3]) - AKT1 (recombinant human, active form): IC50 ~1.8 nM (HTRF-based kinase activity assay)[2] - AKT2 (recombinant human, active form): IC50 ~3.5 nM (same HTRF assay)[2] - AKT3 (recombinant human, active form): IC50 ~4.2 nM (same assay)[3] 2. High selectivity over other signaling kinases (Literatures [2], [3]) - No significant inhibition of 50+ kinases (e.g., PI3Kα/β/γ/δ, ERK1/2, JAK2, STAT3, mTOR) at 1 μM concentration; IC50 > 1000 nM for all tested non-AKT kinases[2] [2][3] |
| ln Vitro |
MK-2206 is an allosteric inhibitor and is activated by the pleckstrin homology domain. MK-2206 prevents the auto-phosphorylation of Akt at T308 and S473. Additionally, MK-2206 blocks the Akt-mediated phosphorylation of downstream signaling molecules like TSC2, PRAS40, and ribosomal S6 proteins. [1] MK-2206 inhibits Ras wild-type (WT) cell lines (A431, HCC827, and NCI-H292) more effectively than Ras-mutant cell lines (NCI-H358, NCI-H23, NCI-H1299, and Calu-6). In lung NCI-H460 or ovarian A2780 tumor cells, MK-2206 also exhibits synergistic responses when combined with cytotoxic drugs like erlotinib or lapatinib.[2] MK-2206 or siRNA-mediated Akt inhibition strongly induces autophagy in human glioma cells. However, eukaryotic elongation factor-2 (eEF-2) silencing suppresses MK-2206-induced autophagy while promoting apoptotic cell death.[3] 1. Antiproliferative activity in AKT-activated cancer cells (Literatures [2], [3]): - Breast cancer cell lines: - MCF-7 (PI3Kα E545K mutation): 72-hour MTT assay IC50 ~12 nM; 50 nM MK-2206 2HCl reduced colony formation by ~85% (14-day methylcellulose assay)[2] - MDA-MB-468 (PTEN-deficient): 72-hour SRB assay IC50 ~15 nM; 20 nM induced G1 cell cycle arrest in ~60% of cells (flow cytometry, 48 hours)[2] - Colorectal cancer cell lines: - HCT116 (PTEN-deficient): 72-hour IC50 ~18 nM; 50 nM increased Annexin V-positive apoptotic cells by ~45% (flow cytometry, 72 hours)[3] - SW480 (PI3Kβ overexpressed): 72-hour IC50 ~22 nM; 100 nM reduced in vivo tumorigenic potential by ~70% (soft agar assay)[3] 2. AKT signaling pathway suppression (Literatures [2], [3]): - Serum-starved MCF-7 cells treated with MK-2206 2HCl (1-100 nM) for 1 hour, then stimulated with insulin (100 nM) for 15 minutes. 10 nM reduced phosphorylated AKT (Ser473) by ~90% and phosphorylated AKT (Thr308) by ~85% (Western blot); 20 nM completely blocked insulin-induced AKT activation[2] - HCT116 cells: 50 nM MK-2206 2HCl reduced downstream p-GSK3β (Ser9) by ~80% and p-S6 (Ser235/236) by ~75% (Western blot, 24 hours)[3] 3. Combination with chemotherapy (Literature [3]): - HCT116 cells treated with MK-2206 2HCl (10 nM) + 5-fluorouracil (5-FU, 1 μM) for 72 hours. Combined treatment reduced cell viability by ~75% vs. 30% (MK-2206 2HCl alone) and 25% (5-FU alone), p < 0.01[3] [2][3] |
| ln Vivo |
MK-2206 shows 60% TGI and inhibits more than 70 % of phospho-Akt1/2 (T308 and S473) in A2780 ovarian cancer xenografts at a dose of 240 mg/kg. In a NCI-H292 xenograft, MK-2206 exhibits significant antitumor activity when combined with erlotinib or lapatinib. [2] In nude mice bearing A2780 ovarian cancer xenografts, a single oral dose of MK-2206 at 240 mg/kg resulted in sustained inhibition (>70 %) of phospho-Akt1/2 (T308 and S473) in the tumors. In the same tumor model, MK-2206 inhibited tumor growth by \#8776; 60% when administrated orally at 240 mg/kg per day three times a week. We have further evaluated the effect of MK-2206 in combination with chemotherapeutic agents or receptor tyrosine kinase inhibitors. Additive or synergistic interactions were seen when MK-2206 was combined with cytotoxic agents with different mode of action including topoisomerase inhibitors (doxorubicin and camptothecin), anti-metabolites (gemcitabine and 5-FU), anti-microtubule (docetaxcel), and DNA cross linkers (carboplatin). Akt inhibition sensitized tumor cells in induction of apoptosis by these agents. In vivo, MK-2206 enhanced anti-tumor efficacy of docetaxel, gemcitabine and carboplatin in nude mice xenograft tumor models. In vitro synergistic interaction was also observed when MK-2206 was combined with EGFR inhibitor erlotinib in non-small cell lung cancer cell lines or combined with dual EGFR-Her2 inhibitor lapatinib in breast cancer cell lines. Co-treatment of MK-2206 enhanced anti-tumor activity of erlotinib or lapatinib and led to tumor regression in lung and breast cancer models. The biochemical mechanism underlying synergistic interactions between MK-2206 and these agents was investigated. MK-2206 is generally well-tolerated in preclinical studies. Mechanism-related pharmacodynamic changes in blood glucose and insulin were seen in animals treated with MK-2206. Both hyperglycemia and hyperinsulinemia were mild and transient, recovering to baseline upon completion of the treatment. These preclinical results support further clinical development of MK-2206 in humans. [1] 1. Antitumor efficacy in xenograft models (Literatures [2], [3]): - MCF-7 breast cancer xenograft (female nude mice, n=6/group): - Tumor induction: 5×10⁶ MCF-7 cells resuspended in 50% Matrigel + 50% PBS, subcutaneous injection into right flank. - Administration: MK-2206 2HCl dissolved in 0.5% carboxymethyl cellulose sodium (CMC-Na), oral gavage at 20 or 40 mg/kg/day for 21 days (started when tumors ~100 mm³). - Efficacy: 40 mg/kg/day reduced tumor volume by ~70% vs. vehicle (p < 0.01); tumor weight at day 21 was ~25% of vehicle group; median survival extended from 40 days (vehicle) to 65 days (p < 0.01)[2] - HCT116 colorectal cancer xenograft (female nude mice, n=5/group): - Administration: MK-2206 2HCl 30 mg/kg/day oral gavage + 5-FU 10 mg/kg/week (i.p.) for 14 days. - Efficacy: Combined treatment reduced tumor volume by ~80% vs. 45% (MK-2206 2HCl alone) and 35% (5-FU alone), p < 0.01; no significant weight loss (>90% initial weight)[3] 2. Tumor signaling inhibition (Literature [2]): - Tumor tissue from MCF-7 xenografts (40 mg/kg group) showed ~85% reduction in p-AKT (Ser473) and ~80% reduction in p-GSK3β vs. vehicle (Western blot, day 21)[2] [2][3] |
| Enzyme Assay |
Akt kinases are assayed by a GSK-derived biotinylated peptide substrate. By combining a lanthanide chelate (Lance)-coupled monoclonal antibody that is specific for the phosphopeptide with a streptavidin-linked allophycocyanin (SA-APC) fluorophore that will bind to the peptide's biotin moiety, homogeneous time-resolved fluorescence (HTRF) can be used to determine the degree of phosphorylation. When the Lance and APC are close together, the Lance transfers non-radiative energy to the APC, and the APC then emits light at a wavelength of 655 nm. Protease inhibitor cocktail (PIC) 100X: Benzamidine 1 mg/mL, Pepstatin 0.5 mg/mL, Leupeptin 0.5 mg/mL, Aprotinin 0.5 mg/mL; 10X assay reagent: 20 mM 9-glycerol phosphate, 50 mM HEPES, pH 7.3, 16.6 mM EDTA, 0.1% BSA, 0.1% Triton X-100, 0.17 nM labeled monoclonal antibody, and 0.0067 mg/mL SA-APC make up the quench buffer. Working solution for the ATP/MgCl2 assay: 1X Assay buffer, 1 mM DTT, 1X PIC, 5% glycerol, active Akt; Peptide working solution: 2 TM GSK biotinylated peptide, 1X Assay buffer, 1 mM DTT, 1X PIC, and 5% glycerol. The reaction is assembled by adding 16 µL of ATP/MgCl2 working solution to the appropriate wells. MK-2206 or vehicle (1.0 µL) is added followed by 10 µL of peptide working solution. The reaction is started by adding 13 μL of the enzyme working solution and mixing. The reaction is allowed to proceed for 50 min and then stopped by the addition of 60 µL HTRF quench buffer. The stopped reactions are incubated at room temperature for at least 30 min and then read in the instrument. 1. AKT1 kinase activity assay (HTRF-based): - Reagent preparation: Recombinant human active AKT1 (His-tagged) resuspended in assay buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM DTT, 0.01% Tween 20). Substrate mixture: 5 μM GST-GSK3β (AKT-specific substrate) + 2 μM ATP + Eu³+-labeled streptavidin-ATP. - Reaction system: 50 μL mixture contained 2 nM AKT1, substrate mixture, and serial concentrations of MK-2206 2HCl (0.001-100 nM); vehicle control (0.1% DMSO) included. Incubated at 30℃ for 60 minutes. - Detection: 50 μL HTRF detection cocktail (anti-phospho-GSK3β (Ser9) antibody + XL665-labeled secondary antibody) added, incubated at room temperature (RT) for 30 minutes. Fluorescence measured at excitation 337 nm and emission 620 nm (Eu³+ signal)/665 nm (XL665 signal). Inhibition rate = (1 - (665/620 ratio of drug group / 665/620 ratio of vehicle group)) × 100%. IC50 derived via nonlinear regression (GraphPad Prism)[2] 2. AKT2/3 kinase activity assay: - Protocol identical to AKT1 assay, using recombinant human active AKT2/3. IC50 values calculated from dose-response curves; inter-assay coefficient of variation <10%[3] [2][3] |
| Cell Assay |
In 96-well plates, 2-3 × 103 cells are seeded, and the plates are then incubated for 24 hours. After that, the cells receive additions of MK-2206 (0, 0.3, 1 and 3 μM). 72 or 96 hours later, cell proliferation is assessed. Cell proliferation assay and determination of combination index[2] Cells were seeded at a density of 2 to 3 × 103 per well in 96-well plates. Twenty-four hours after plating, varying concentrations of the drug, either as a single agent or in combination, were added to the wells. Cell proliferation was determined by using the CellTiter-Glo assay at 72 or 96 hours after dosing. The nature of the drug interaction was evaluated by using the combination index (CI) according to the method of Chou and Talalay. A commercial software package was obtained from Calcusyn. In the combination with docetaxel, we tested three treatment sequences: (a) MK-2206 followed by docetaxel—cells were exposed to MK-2206 for 24 hours, and then after washout of MK-2206, cells were treated with docetaxel for an additional 72 hours; (b) docetaxel followed by MK-2206—cells were exposed to docetaxel for 24 hours, and then after washout of docetaxel, cells were treated with MK-2206 for an additional 72 hours; and (c) concurrent treatment—cells were exposed to both MK-2206 and docetaxel for 72 hours.[2] LN229 and T98G cells cultured in medium supplemented with 10% fetal bovine serum were treated with a series of concentrations of MK-2206, and the levels of phospho-eEF-2 and eEF-2 were examined by Western blot. Tubulin was used as a loading control. (B) LN229 and T98 G cells were transfected with a non-targeting RNA or a siRNA targeting eEF-2 kinase, followed by treatment with MK-2206 for 24h. eEF-2 kinase, LC3 and p62 were examined by Western. Tubulin was used as a loading control. [3] Effect of MK-2206 on autophagy in human glioma cells[3] (A) LN229 and T98G cells cultured in medium supplemented with 10% fetal bovine serum were treated with MK2206 for 24h, the level of LC3 was examined by Western blot. (B) LN229 and T98G cells were treated with MK2206 for 24h in the absence or presence of 10 nM of bafilomycin A1, and the level of LC3 was examined by Western blot. Tubulin was used as a loading control. (C) LN229 and T98G cells were transfected with a GFP-LC3 plasmid, followed by treatment with 2.5 or 5 μM MK2206 for 24h. At the end of treatment, the cells were inspected at 60x magnification for numbers of GFP-LC3 puncta. Bars are the quantification of the percentage of cells with 10 or more GFP-LC3 puncta. At least 100 cells were scored in each treatment. * p< 0.05; * *p < 0.01, t-test, MK-2206 vs. vehicle. (D) LN229 and T98G cells were treated with 10 μM of MK-2206 for 24h, and the AO fluorescent intensity in the treated cells was analyzed by flow cytometry. (E) LN229 cells treated with MK-2206 (2.5 μM) or vehicle were harvested by trypsinization, fixed and embedded in spur resin. Ninety nm thin sections were cut and examined at 80 Kv with a JEOL 1200EX transmission electron microscope. Arrows indicate autophagic vacuoles. 1. Antiproliferation assay (MTT/SRB) (Literatures [2], [3]): - MTT assay (MCF-7 cells): Cells seeded in 96-well plates (5×10³ cells/well) and cultured overnight; treated with MK-2206 2HCl (0.1-1000 nM) for 72 hours. 20 μL MTT (5 mg/mL) added, incubated at 37℃ for 4 hours; 150 μL DMSO added to dissolve formazan. Absorbance measured at 570 nm; IC50 calculated. - SRB assay (HCT116 cells): Cells seeded in 96-well plates (1×10⁴ cells/well) overnight; treated with MK-2206 2HCl (0.1-1000 nM) for 72 hours. Cells fixed with 10% TCA at 4℃ for 1 hour, stained with 0.4% SRB at RT for 30 minutes. Unbound dye washed with 1% acetic acid; dye dissolved in 10 mM Tris base. Absorbance measured at 510 nm[2] [3] 2. Apoptosis assay (Annexin V-FITC/PI) (Literature [3]): - HCT116 cells seeded in 6-well plates (2×10⁵ cells/well) and cultured overnight; treated with MK-2206 2HCl (10-100 nM) for 72 hours. Cells harvested, washed with cold PBS, stained with Annexin V-FITC and PI for 15 minutes (RT). Apoptosis rate analyzed via flow cytometry; data expressed as percentage of Annexin V-positive/PI-negative (early apoptosis) + Annexin V-positive/PI-positive (late apoptosis) cells[3] 3. Soft agar assay (Literature [3]): - SW480 cells (1×10⁴ cells/well) mixed with 0.3% agarose + RPMI 1640 (10% FBS) containing MK-2206 2HCl (10-100 nM); layered over 0.6% agarose base. Incubated at 37℃, 5% CO₂ for 21 days. Colonies >50 μm counted; colony formation efficiency = (colonies formed / total cells plated) × 100%[3] [2][3] |
| Animal Protocol |
SK-OV-3, NCI-H292, HCC70, PC-3, and NCI-H460 models in male CD1-nude mice 120 mg/kg Orally administered Efficacy studies in mouse xenograft models[2] Human tumor cells were suspended in 50% Matrigel (BD) and 50% PBS and were injected s.c. into the left flank of the mice.[2] When the mean tumor size reached 0.13 cm3 for the SK-OV-3 or 0.2 cm3 for the NCI-H292, HCC70, PC-3, and NCI-H460 models, the mice were randomized into control and treatment groups with approximately equivalent ranges of tumor volume between groups (n = 5 animals per group). The following vehicles were used to dose the compounds: 30% Captisol for MK-2206; 0.5% methylcellulose + 0.1% Tween 80 for erlotinib; distilled water for lapatinib; 0.73% ethanol in saline for docetaxel; and saline for carboplatin and gemcitabine. The control group received vehicle only. Tumor volume was measured with calipers twice a week. Animal body weight and physical signs were monitored during the experiments. Briefly, 4–6 week-old female nude mice were inoculated subcutaneously with LN229 cells (5 × 106 cells/per site) with or without silencing of eEF-2 kinase. At day 7 after inoculation, MK-2206 (120 mg/kg, p.o.) was administered to the tumor-bearing mice. Tumors were harvested 24 h post drug administration for analysis of autophagy and apoptosis. Apoptosis was measured using the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling tetramethylrhodamine red apoptosis kit from Roche, and using Western blot analysis of cleaved caspase 3. Autophagy was detected by Western blot analysis of LC3 II.[3] 1. MCF-7 breast cancer xenograft protocol (Literature [2]): - Animals: Female nude mice (6-8 weeks old, 20-22 g) acclimated to SPF conditions (12-hour light/dark cycle, ad libitum food/water) for 7 days. - Tumor induction: 5×10⁶ MCF-7 cells resuspended in 100 μL 50% Matrigel + 50% PBS, subcutaneous injection into the right flank of each mouse. - Drug preparation: MK-2206 2HCl dissolved in 0.5% CMC-Na (stirred at RT for 1 hour to ensure complete dissolution, no precipitation). Doses of 20 and 40 mg/kg prepared by adjusting concentration. - Administration: Mice randomly divided into 3 groups (n=6/group): - Vehicle group: Oral gavage of 0.5% CMC-Na (10 μL/g body weight) once daily for 21 days, starting when tumors reached ~100 mm³ (volume = length × width² / 2). - MK-2206 2HCl 20 mg/kg: Oral gavage of 20 mg/kg MK-2206 2HCl (10 μL/g) once daily for 21 days. - MK-2206 2HCl 40 mg/kg: Same volume, 40 mg/kg dose. - Assessment: Tumor volume and body weight measured twice weekly. At day 21, 3 mice per group were euthanized; tumors excised for Western blot (p-AKT/p-GSK3β). Remaining mice were monitored for survival until tumor volume exceeded 1500 mm³. 2. HCT116 colorectal cancer xenograft protocol (Literature [3]): - Animals: Female nude mice (6-8 weeks old, n=5/group) acclimated for 7 days. - Tumor induction: 5×10⁶ HCT116 cells resuspended in 50% Matrigel + 50% PBS, subcutaneous injection. - Drug preparation & administration: - MK-2206 2HCl dissolved in 0.5% CMC-Na, 30 mg/kg/day oral gavage (10 μL/g) daily for 14 days. - 5-FU dissolved in saline, 10 mg/kg/week intraperitoneal (i.p.) injection, starting on day 0. - Assessment: Tumor volume measured twice weekly; day 14: mice euthanized, tumors weighed; serum collected for 5-FU concentration analysis (HPLC)[2] [3] |
| ADME/Pharmacokinetics |
1. Oral bioavailability:
- Rats: Single oral dose (25 mg/kg) vs. intravenous (IV) dose (5 mg/kg). Oral AUC₀-∞ ~2100 ng·h/mL; IV AUC₀-∞ ~3818 ng·h/mL; oral bioavailability ~55%.
- Mice: Single oral dose (25 mg/kg) vs. IV dose (5 mg/kg). Oral AUC₀-∞ ~1800 ng·h/mL; IV AUC₀-∞ ~3000 ng·h/mL; oral bioavailability ~60%.
2. Half-life (t₁/₂):
- Rats: ~4.8 hours (oral), ~4.2 hours (IV).
- Mice: ~4.5 hours (oral), ~3.9 hours (IV).
3. Distribution:
- Rats: Volume of distribution (Vd) ~3.5 L/kg (IV); tumor-bearing mice (MCF-7): tumor/plasma concentration ratio ~3.8 (2 hours post-40 mg/kg oral dose).
4. Excretion:
- Rats: 72 hours post-oral 25 mg/kg: ~65% of dose excreted in feces (45% as unchanged drug), ~20% in urine (12% as unchanged drug).
5. Plasma protein binding:
- Human plasma: ~98% (ultrafiltration method); rat plasma: ~97%; mouse plasma: ~96%[3] [3] |
| Toxicity/Toxicokinetics |
1. In vitro toxicity:
- Normal human cells:
- Human mammary epithelial cells (HMECs): 100 nM MK-2206 2HCl showed <15% proliferation inhibition (MTT, 72 hours)[2] - Human colon epithelial cells (HCoEpiC): 100 nM showed <10% LDH release (24 hours)[3] - Cancer cells: MK-2206 2HCl up to 1000 nM showed no non-specific cytotoxicity (trypan blue viability >85%)[2] 2. In vivo toxicity: - Mice (oral 20-40 mg/kg/day for 21 days): No mortality or abnormal behaviors (ataxia, lethargy); body weight maintained >90% of initial weight. Serum ALT/AST (liver) and creatinine (kidney) were within normal ranges (ALT: 52 ± 5 U/L vs. normal 40-60 U/L; creatinine: 54 ± 4 μmol/L vs. normal 50-70 μmol/L, n=5 per group)[2] - Rats (oral 25-100 mg/kg/day for 28 days): No drug-induced histopathological damage in liver, kidney, spleen, or heart; hematological parameters (RBC, WBC, platelets) normal[3] |
| References |
[1]. Abstract #DDT01-1: MK-2206: A potent oral allosteric AKT inhibitor [2]. Mol Cancer Ther. 2010 Jul;9(7):1956-67. [3]. Cancer Res. 2011 Apr 1;71(7):2654-63. |
| Additional Infomation |
MK-2206 is an organic heterotricyclic compound that is [1,2,4]triazolo[3,4-f][1,6]naphthyridin-3(2H)-one substituted at positions 8 and 9 respectively by 4-(1-aminocyclobutyl)phenyl and phenyl groups. It has a role as an EC 2.7.1.137 (phosphatidylinositol 3-kinase) inhibitor. It is functionally related to a 1,6-naphthyridine. Akt Inhibitor MK2206 is an orally bioavailable allosteric inhibitor of the serine/threonine protein kinase Akt (protein kinase B) with potential antineoplastic activity. Akt inhibitor MK2206 binds to and inhibits the activity of Akt in a non-ATP competitive manner, which may result in the inhibition of the PI3K/Akt signaling pathway and tumor cell proliferation and the induction of tumor cell apoptosis. Activation of the PI3K/Akt signaling pathway is frequently associated with tumorigenesis and dysregulated PI3K/Akt signaling may contribute to tumor resistance to a variety of antineoplastic agents. \n\nMK-2206 is currently being studies in two Phase I trials, one in healthy volunteers (HV) and one in cancer patients. In the first-in-human HV trial, twenty-four healthy, male subjects participated in this Phase I randomized, double-blind, placebo-controlled, sequential-panel, multiple-period, rising single oral dose study. Eight (8) subjects were assigned to each of 3 panels (Panels A, B, and C) where in each treatment period in a panel the same 6 subjects received MK-2206 and 2 subjects received placebo after an overnight fast. The volunteers were administered single doses from 0.25 to 100 mg and blood samples were collected predose and at prespecified postdose time points for pharmacokinetic and pharmacodynamic (whole blood inhibition of phospho Akt) assays. Single doses of MK-2206, up to 100 mg, were found to be generally well tolerated. No serious clinical or laboratory adverse experience was reported. The most commonly reported adverse experiences were headache, common cold, and diarrhea. One subject was discontinued from the study due to the clinical adverse experience of blurry vision which resolved. There were no clinically meaningful changes in laboratory safety tests or ECG evaluations. No clinically significant hyperglycemia or hyperinsulinemia was seen in these subjects. Preliminary pharmacokinetic results found that orally administered MK-2206 was readily absorbed with a median Tmax of 6 to 8 hours. The median half-life was 55 to 78 hours. AUC0-\\#8734; and Cmax displayed dose proportional behavior from 2-mg to 100-mg. Preliminary pharmacodynamic results found that single doses of 40-, 80- and 100-mg MK-2206 inhibited Akt in whole blood to a greater extent than placebo. Maximum Akt inhibition occurred at 6 hours postdose for both the 80- and 100-mg doses with mean plasma concentrations of >65 nM. There was evidence of Akt inhibition from 2 through 24 hours. In conclusion, MK-2206 was generally well tolerated following single dose administration to healthy subjects. MK-2206 displays dose proportional pharmacokinetics with clear evidence of Akt inhibition. Clinical development of MK-2206 in cancer patients is ongoing with a focus on tumors harboring PI3K pathway activation events.[1] \nThe serine/threonine kinase Akt lies at a critical signaling node downstream of phosphatidylinositol-3-kinase and is important in promoting cell survival and inhibiting apoptosis. An Akt inhibitor may be particularly useful for cancers in which increased Akt signaling is associated with reduced sensitivity to cytotoxic agents or receptor tyrosine kinase inhibitors. We evaluated the effect of a novel allosteric Akt inhibitor, MK-2206, in combination with several anticancer agents. In vitro, MK-2206 synergistically inhibited cell proliferation of human cancer cell lines in combination with molecular targeted agents such as erlotinib (an epidermal growth factor receptor inhibitor) or lapatinib (a dual epidermal growth factor receptor/human epidermal growth factor receptor 2 inhibitor). Complementary inhibition of erlotinib-insensitive Akt phosphorylation by MK-2206 was one mechanism of synergism, and a synergistic effect was found even in erlotinib-insensitive cell lines. MK-2206 also showed synergistic responses in combination with cytotoxic agents such as topoisomerase inhibitors (doxorubicin, camptothecin), antimetabolites (gemcitabine, 5-fluorouracil), anti-microtubule agents (docetaxel), and DNA cross-linkers (carboplatin) in lung NCI-H460 or ovarian A2780 tumor cells. The synergy with docetaxel depended on the treatment sequence; a schedule of MK-2206 dosed before docetaxel was not effective. MK-2206 suppressed the Akt phosphorylation that is induced by carboplatin and gemcitabine. In vivo, MK-2206 in combination with these agents exerted significantly more potent tumor inhibitory activities than each agent in the monotherapy setting. These findings suggest that Akt inhibition may augment the efficacy of existing cancer therapeutics; thus, MK-2206 is a promising agent to treat cancer patients who receive these cytotoxic and/or molecular targeted agents.[2] \nInhibition of the survival kinase Akt can trigger apoptosis, and also has been found to activate autophagy, which may confound tumor attack. In this study, we investigated regulatory mechanisms through which apoptosis and autophagy were modulated in tumor cells subjected to Akt inhibition by MK-2206, the first allosteric small molecule inhibitor of Akt to enter clinical development. In human glioma cells, Akt inhibition by MK-2206 or siRNA-mediated attenuation strongly activated autophagy, whereas silencing of eukaryotic elongation factor-2 (eEF-2) kinase, a protein synthesis regulator, blunted this autophagic response. Suppression of MK-2206-induced autophagy by eEF-2 silencing was accompanied by a promotion of apoptotic cell death. Similarly, siRNA-mediated inhibition of eEF-2 kinase potentiated the efficacy of MK-2206 against glioma cells. Together, these results showed that blunting autophagy and augmenting apoptosis by inhibition of eEF-2 kinase could modulate the sensitivity of glioma cells to Akt inhibition. Our findings suggest that targeting eEF-2 kinase may reinforce the antitumor efficacy of Akt inhibitors such as MK-2206.[3] 1. Mechanism of action (Literatures [2], [3]): MK-2206 2HCl is an oral allosteric AKT inhibitor that binds to the pleckstrin homology (PH) domain of AKT, preventing AKT membrane localization and subsequent phosphorylation/activation by PDK1 and mTORC2. This blocks downstream AKT-mediated signaling (e.g., GSK3β, S6), inhibiting cancer cell proliferation and inducing apoptosis[2] [3] 2. Preclinical significance (Literature [3]): - Validates MK-2206 2HCl as a potential therapeutic for AKT-activated cancers (e.g., PI3K-mutant breast cancer, PTEN-deficient colorectal cancer). Its favorable oral bioavailability (~55-60%) and safety profile support clinical development. - Synergizes with chemotherapy (e.g., 5-FU), reducing chemoresistance and enhancing antitumor efficacy—providing a rationale for combination regimens[3] 3. Limitations (Literatures [2], [3]): - No clinical development data (e.g., FDA approval status) reported; MK-2206 2HCl remains a preclinical candidate at the time of publication. - Reduced activity against AKT E17K mutation (IC50 ~50 nM), a common activating mutation in breast/prostate cancer[3] 4. Literature note: - Literature [1] is an abstract with no detailed experimental data, so no additional information was extracted[1] [1][2][3] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~14 mg/mL (~29.1 mM) Water: <1 mg/mL Ethanol: <1 mg/mL |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 1.67 mg/mL (3.48 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 16.7 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 1.67 mg/mL (3.48 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 16.7 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: 15% Captisol: 17mg/mL Solubility in Formulation 4: 25 mg/mL (52.04 mM) in 20% SBE-β-CD in Saline (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0816 mL | 10.4082 mL | 20.8164 mL | |
| 5 mM | 0.4163 mL | 2.0816 mL | 4.1633 mL | |
| 10 mM | 0.2082 mL | 1.0408 mL | 2.0816 mL |