Physicochemical Properties
| Molecular Formula | C20H18N6O3 |
| Molecular Weight | 390.3953 |
| Exact Mass | 390.144 |
| CAS # | 2271081-58-2 |
| PubChem CID | 138454765 |
| Appearance | Light yellow to yellow solid powder |
| LogP | 3.7 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 29 |
| Complexity | 597 |
| Defined Atom Stereocenter Count | 0 |
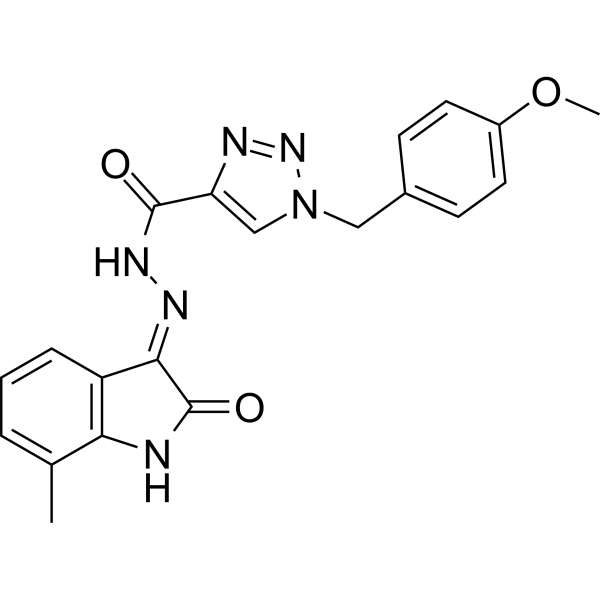
| SMILES | O([H])C1=C(C2=C([H])C([H])=C([H])C(C([H])([H])[H])=C2N1[H])/N=N/C(C1=C([H])N(C([H])([H])C2C([H])=C([H])C(=C([H])C=2[H])OC([H])([H])[H])N=N1)=O |
| InChi Key | KACGMLSYPGROFF-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C20H18N6O3/c1-12-4-3-5-15-17(12)21-20(28)18(15)23-24-19(27)16-11-26(25-22-16)10-13-6-8-14(29-2)9-7-13/h3-9,11,21,28H,10H2,1-2H3 |
| Chemical Name | N-[(2-hydroxy-7-methyl-1H-indol-3-yl)imino]-1-[(4-methoxyphenyl)methyl]triazole-4-carboxamide |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
MARK4 inhibitor 1 targets microtubule affinity-regulating kinase 4 (MARK4) (IC50 = 0.08 μM for human MARK4 kinase activity; Ki = 0.04 μM, competitive inhibition mode) [1] MARK4 inhibitor 1 shows high selectivity over other kinases (MARK1-3, CDK2, EGFR, VEGFR2; IC50 > 10 μM; selectivity index > 125 vs. MARK4) [1] |
| ln Vitro |
MARK4 inhibitor 1 (compound 9g; 0-200 µM; 24-48 hours) suppresses the growth and migration of cells [1]. Compound 9g; 24, also known as MARK4 Inhibitor 1, causes these sodium phosphates with IC50 values of 6.22 μM. - MARK4 kinase inhibitory activity: MARK4 inhibitor 1 potently and selectively inhibited recombinant human MARK4 kinase activity in a dose-dependent manner, with IC50 = 0.08 μM and Ki = 0.04 μM. Kinetic analysis confirmed it competes with ATP for binding to the MARK4 ATP-binding pocket [1] - Antiproliferative activity: The compound inhibited proliferation of various cancer cell lines with high MARK4 expression. IC50 values were 0.9 μM (MCF-7, breast cancer), 1.2 μM (A549, lung cancer), 1.5 μM (HCT116, colon cancer), and 0.7 μM (HeLa, cervical cancer). It had minimal cytotoxicity to normal human foreskin fibroblasts (NHF, IC50 > 20 μM) [1] - Induction of apoptosis: Flow cytometry analysis showed that MARK4 inhibitor 1 (1 μM, 2 μM) induced apoptosis in MCF-7 cells. At 2 μM, apoptotic rate increased from 4% (control) to 38%, with upregulated Bax/Bcl-2 ratio (3.2-fold vs. control) and activated cleaved caspase-3 (2.8-fold vs. control) detected by western blot [1] - Inhibition of cell migration and invasion: MARK4 inhibitor 1 (0.5-2 μM) suppressed migration and invasion of A549 and HeLa cells. At 2 μM, migration rate was reduced by 65% (A549) and 70% (HeLa), and invasion rate by 68% (A549) and 72% (HeLa) compared to control, as determined by Transwell assays [1] - Disruption of microtubule dynamics: Immunofluorescence staining revealed that MARK4 inhibitor 1 (2 μM) disrupted microtubule organization in MCF-7 cells, with reduced microtubule stability. It also inhibited MARK4-mediated phosphorylation of microtubule-associated protein tau (p-tau), with a 62% reduction in p-tau levels at 2 μM [1] - Cell cycle arrest: Flow cytometry analysis showed that MARK4 inhibitor 1 (1-2 μM) induced G2/M phase arrest in A549 cells. At 2 μM, G2/M phase cells increased from 14% (control) to 41% [1] |
| ln Vivo |
- Antitumor efficacy in MCF-7 breast cancer xenograft model: In nude mice bearing MCF-7 xenografts, oral administration of MARK4 inhibitor 1 (15 mg/kg, 30 mg/kg, once daily for 21 days) resulted in tumor growth inhibition rates of 56% and 68% respectively. Tumor weight was reduced by 65% (30 mg/kg) compared to vehicle control. No significant body weight loss (< 7%) was observed [1] - Mechanism in vivo: Tumor tissues from treated mice (30 mg/kg) showed reduced MARK4 kinase activity (62% inhibition), decreased p-tau levels (58% reduction), and increased apoptotic cells (3.5-fold vs. control) as detected by TUNEL staining. Western blot confirmed upregulated Bax and cleaved caspase-3, and downregulated Bcl-2 [1] |
| Enzyme Assay |
- MARK4 kinase activity assay: Recombinant human MARK4 kinase domain was mixed with ATP (10 μM), fluorescently labeled peptide substrate (derived from tau protein), and MARK4 inhibitor 1 at gradient concentrations (0.001-1 μM) in kinase buffer (pH 7.4). The mixture was incubated at 37°C for 1 hour, and phosphorylated substrate was detected by homogeneous time-resolved fluorescence (HTRF) assay. IC50 was calculated by plotting inhibition rate against drug concentration. Kinetic analysis with varying ATP concentrations confirmed competitive inhibition mode [1] - Kinase selectivity assay: Recombinant MARK1-3, CDK2, EGFR, VEGFR2, and other kinases were separately mixed with their corresponding substrates, ATP, and MARK4 inhibitor 1 (10 μM) in kinase buffer. After 37°C incubation for 1 hour, enzyme activity was detected by HTRF assay to evaluate selectivity [1] |
| Cell Assay |
Cell Proliferation Assay [1] Cell Types: MCF-7, MDA-MB-435s and HepG2 Cell Tested Concentrations: 0-200 μM Incubation Duration: 24 and 48 hour Experimental Results: diminished viability of these cells. Concentration dependent manner. - Cell viability assay: Cancer cells (MCF-7, A549, HCT116, HeLa) and NHF cells were seeded into 96-well plates at 5×10³ cells/well, treated with MARK4 inhibitor 1 (0.01-20 μM) for 72 hours. Cell viability was measured by tetrazolium salt-based assay, and IC50 values were calculated [1] - Apoptosis assay: MCF-7 cells were seeded into 6-well plates, treated with MARK4 inhibitor 1 (1 μM, 2 μM) for 48 hours. Apoptotic cells were quantified by Annexin V-FITC/PI staining and flow cytometry. For protein analysis, cells were lysed, and Bax, Bcl-2, cleaved caspase-3, and GAPDH proteins were detected by western blot [1] - Cell cycle assay: A549 cells were treated with MARK4 inhibitor 1 (1 μM, 2 μM) for 24 hours, fixed, stained with propidium iodide, and cell cycle distribution was analyzed by flow cytometry [1] - Migration and invasion assay: A549 and HeLa cells were seeded into Transwell chambers (migration) or Matrigel-coated Transwell chambers (invasion) with MARK4 inhibitor 1 (0.5-2 μM). After 24 hours (migration) or 48 hours (invasion), cells that migrated/invaded to the lower chamber were fixed, stained, and counted. Inhibition rate was calculated relative to control [1] - Immunofluorescence assay for microtubules: MCF-7 cells were grown on coverslips, treated with MARK4 inhibitor 1 (2 μM) for 12 hours, fixed, stained with α-tubulin antibody and DAPI. Fluorescence images were captured to observe microtubule organization [1] - Western blot for p-tau: MCF-7 cells were treated with MARK4 inhibitor 1 (0.5-2 μM) for 24 hours, lysed, and p-tau (Ser262) and total tau proteins were detected by western blot. Band intensities were quantified by densitometry [1] |
| Animal Protocol |
- MCF-7 breast cancer xenograft model: Female nude mice (6-8 weeks old) were subcutaneously injected with MCF-7 cells (5×10⁶ cells/mouse). When tumors reached ~100 mm³, mice were randomly divided into vehicle control, 15 mg/kg, and 30 mg/kg MARK4 inhibitor 1 groups (n=6 per group) [1] - Drug formulation and administration: MARK4 inhibitor 1 was dissolved in a mixture of DMSO, PEG400, and sterile water (volume ratio 1:3:6) to prepare oral suspension. Mice were administered orally once daily for 21 days, with the control group receiving equal volume of the vehicle mixture [1] - Tumor monitoring and tissue analysis: Tumor volume was measured every 3 days (volume = length × width² / 2), and body weight was recorded weekly. At the end of treatment, mice were sacrificed, tumors were excised, weighed, and stored at -80°C. Tumor lysates were used for western blot analysis; tumor sections were subjected to TUNEL staining to detect apoptotic cells [1] |
| ADME/Pharmacokinetics |
- Plasma protein binding: MARK4 inhibitor 1 had a plasma protein binding rate of 91.2 ± 1.8% in human plasma, determined by equilibrium dialysis [1] - In vitro metabolic stability: The compound exhibited moderate metabolic stability in human liver microsomes, with a half-life (t1/2) of 4.5 hours and metabolic clearance rate of 0.48 mL/min/mg protein [1] - In vivo pharmacokinetics in mice: After a single oral dose of 30 mg/kg, Cmax was 8.3 μM, AUC₀₋₂₄h was 46.7 μM·h, elimination half-life (t1/2) was 4.3 hours, and oral bioavailability (F) was 48.5% [1] |
| Toxicity/Toxicokinetics |
- Acute toxicity: Mice showed no mortality or obvious toxicity symptoms (weight loss, lethargy) after a single oral dose of MARK4 inhibitor 1 up to 300 mg/kg, with maximum tolerated dose (MTD) > 300 mg/kg [1] - Subacute toxicity: In mice treated with MARK4 inhibitor 1 (30 mg/kg, oral, once daily for 28 days), no significant changes were observed in body weight, blood routine parameters (WBC, RBC, PLT), or liver/kidney function indices (ALT, AST, creatinine, urea nitrogen). Histopathological examination of major organs (heart, liver, spleen, lungs, kidneys) revealed no abnormal lesions [1] |
| References |
[1]. Design and development of Isatin-triazole hydrazones as potential inhibitors of microtubule affinity-regulating kinase 4 for the therapeutic management of cell proliferation and metastasis. Eur J Med Chem. 2019 Feb 1;163:840-852. |
| Additional Infomation |
- Chemical classification: MARK4 inhibitor 1 is a small-molecule inhibitor, belonging to the isatin-triazole hydrazone derivative class [1] - Mechanism of action: The compound binds to the ATP-binding pocket of MARK4, competitively inhibiting its serine/threonine kinase activity. This leads to G2/M cell cycle arrest, disrupted microtubule dynamics (via reduced tau phosphorylation), induction of apoptosis (upregulating Bax/Bcl-2 ratio and activating caspase-3), and inhibition of tumor cell migration and invasion [1] - Target background: MARK4 is a member of the MARK kinase family, involved in regulating microtubule stability, cell cycle progression, and cell migration. Aberrant overexpression or activation of MARK4 is associated with the development and metastasis of various cancers (e.g., breast, lung, colon cancer) [1] - Therapeutic potential: MARK4 inhibitor 1 is a potent, selective, and orally bioavailable MARK4 inhibitor, showing promising efficacy in inhibiting tumor cell proliferation and metastasis with favorable safety profiles, making it a potential candidate for cancer therapy [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~5 mg/mL (~12.81 mM) |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5615 mL | 12.8074 mL | 25.6148 mL | |
| 5 mM | 0.5123 mL | 2.5615 mL | 5.1230 mL | |
| 10 mM | 0.2561 mL | 1.2807 mL | 2.5615 mL |