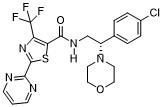
Lu AF27139 a novel rodent-active and CNS-penetrant P2X7 receptor antagonist, which is highly selective and potent against rat, mouse, and human forms of the receptors.
Physicochemical Properties
| Molecular Formula | C21H19CLF3N5O2S |
| Molecular Weight | 497.9211 |
| Exact Mass | 497.09 |
| Elemental Analysis | C, 50.66; H, 3.85; Cl, 7.12; F, 11.45; N, 14.07; O, 6.43; S, 6.44 |
| CAS # | 2097117-06-9 |
| PubChem CID | 129099048 |
| Appearance | White to off-white solid powder |
| LogP | 3.3 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 10 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 33 |
| Complexity | 644 |
| Defined Atom Stereocenter Count | 1 |
| SMILES | C1COCCN1[C@H](CNC(=O)C2=C(N=C(S2)C3=NC=CC=N3)C(F)(F)F)C4=CC=C(C=C4)Cl |
| InChi Key | FGPQIEDRTXLBES-OAHLLOKOSA-N |
| InChi Code | InChI=1S/C21H19ClF3N5O2S/c22-14-4-2-13(3-5-14)15(30-8-10-32-11-9-30)12-28-19(31)16-17(21(23,24)25)29-20(33-16)18-26-6-1-7-27-18/h1-7,15H,8-12H2,(H,28,31)/t15-/m1/s1 |
| Chemical Name | (S)-N-(2-(4-chlorophenyl)-2-morpholinoethyl)-2-(pyrimidin-2-yl)-4-(trifluoromethyl)thiazole-5-carboxamide |
| Synonyms | Lu AF-27139Lu AF27139 Lu AF 27139LuAF-27139 LuAF27139 LuAF 27139 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Lu AF27139 targets human P2X7 receptor (Ki = 0.8 nM; functional IC50 for Ca²⁺ influx = 3.2 nM) [1] Lu AF27139 targets rat P2X7 receptor (Ki = 1.2 nM; functional IC50 for Ca²⁺ influx = 4.5 nM) [1] Lu AF27139 targets mouse P2X7 receptor (Ki = 1.5 nM; functional IC50 for Ca²⁺ influx = 5.1 nM) [1] |
| ln Vitro |
Lu AF27139 (compound 1) (10-200 nM) exhibits dose-responsive inhibition of 100 μM BzATP-induced currents in HEK293 cells transfected with rat P2X7R, with an IC50 of 66 nM [1]. In primary rat microglia, Lu AF27139 (compound 1) (100 nM) inhibits 300 μM BzATP-induced currents with an 80% inhibition rate at 100 nM dose [1]. Lu AF27139 (compound 1) has an IC50 of 38 ± 2.5 nM, which inhibits the release of IL-1β from THP-1 cells when they are primed with LPS and when BzATP is added [1]. Compound 1, Lu AF27139, has an IC50 of 38 ± 38 in rats and inhibits the release of IL-1β in primary cortical microglia of mice and rats that have been primed with LPS and stimulated with 1 mM BzATP in a concentration-dependent manner. 19 nM, and in mice, its IC50 was 26 ± 6 nM[1]. 1. Shows high selectivity for P2X7 receptor over other P2X subtypes (P2X1-P2X6): IC50 > 1000 nM for P2X1, P2X2, P2X3, P2X4, P2X5, P2X6 receptors in functional assays [1] 2. Inhibits ATP-induced Ca²⁺ influx in HEK293 cells expressing human/rat/mouse P2X7 receptors in a concentration-dependent manner, with IC50 values as reported in the Target section [1] 3. Blocks ATP-induced YO-PRO-1 dye uptake (a marker of P2X7 receptor pore formation) in human P2X7-expressing HEK293 cells (IC50 = 2.8 nM) and rat primary microglia (IC50 = 3.9 nM) [1] 4. Reduces LPS-primed, ATP-induced IL-1β release in human peripheral blood mononuclear cells (PBMCs) (IC50 = 4.3 nM) and rat primary macrophages (IC50 = 5.7 nM) [1] 5. No significant inhibition of other ion channels (e.g., TRPV1, Nav1.7) or GPCRs (e.g., CB1, μ-opioid receptor) at concentrations up to 10 μM [1] |
| ln Vivo |
Compound 1, Lu AF27139, when taken orally at doses of 3, 10, and 100 mg/kg, decreases the release of IL-1β in the frontal cortex of rats and mice when LPS is administered intracerebroventricularly (icv) and when BzATP is triggered [1]. Assessing the pharmacokinetic (PK) properties of compound 1 (Lu AF27139) in rats [1]. Dose Cu, plasma (nM)a Cu, brain (nM)a Cu, spinal cord (nM)a (mg/kg, po) (1 h) (2 h) (1 h) (2 h) (1 h) 2 h) T1 22.4 ± 4.2 22.8 ± 10 5.4 ± 2.6 6.4 ± 2.0 5.20 ± 0.80 10.0 ± 2.0 a: The concentrations of Lu AF27139 in rats' free plasma, brain, and spinal cord were calculated using the formula (Ct*fu), where Ct is the total tissue (plasma, brain, or spinal cord) drug concentration, and fu is the unbound fraction of these tissues ascertained by ex vivo equilibrium dialysis. The values from n = 3 animals are expressed as mean ± SEM. Fu = 0.02 ± 0.00 for plasma, 0.07 ± 0.03 for spinal cord, and 0.09 ± 0.03 for brain. The results for experiments with n ≥ 3 are presented as mean ± SEM. 1. In rats, oral administration of Lu AF27139 (3 mg/kg) results in brain concentrations of ~2.9 μM (at 1 hour post-dose), which exceeds the in vitro IC50 for P2X7 inhibition [1] 2. In mice with LPS+ATP-induced peritoneal inflammation, oral Lu AF27139 (1, 3, 10 mg/kg) dose-dependently reduces IL-1β levels in peritoneal fluid (inhibition rates: 32%, 58%, 75% at 1, 3, 10 mg/kg, respectively) [1] 3. In rats subjected to spinal nerve ligation (neuropathic pain model), oral Lu AF27139 (3, 10 mg/kg) significantly reduces mechanical allodynia (paw withdrawal threshold increased by ~40% and ~65% at 3 and 10 mg/kg, respectively) [1] 4. Crosses the blood-brain barrier (BBB) in both rats and mice, with brain/plasma concentration ratios of ~0.8 (rats) and ~0.7 (mice) at 1 hour post-oral administration of 3 mg/kg [1] |
| Enzyme Assay |
1. P2X7 receptor radioligand binding assay: Membranes from HEK293 cells expressing human/rat/mouse P2X7 receptors are incubated with a tritiated P2X7-selective ligand. Lu AF27139 is added at serial concentrations (0.1 nM-10 μM) to compete for binding sites. After incubation at 25°C for 60 minutes, unbound ligand is removed by filtration, and bound radioactivity is measured. Ki values are calculated using nonlinear regression analysis [1] 2. P2X7 functional Ca²⁺ influx assay: HEK293 cells expressing human/rat/mouse P2X7 receptors are loaded with a fluorescent Ca²⁺ indicator. Cells are preincubated with Lu AF27139 (0.1 nM-10 μM) for 15 minutes, then stimulated with ATP (100 μM) to activate P2X7 receptors. Fluorescence intensity is recorded over 5 minutes, and IC50 values are determined based on the inhibition of peak fluorescence response [1] |
| Cell Assay |
1. YO-PRO-1 dye uptake assay: Human P2X7-expressing HEK293 cells or rat primary microglia are seeded in 96-well plates. Cells are preincubated with Lu AF27139 (0.1 nM-10 μM) for 20 minutes, then treated with ATP (300 μM) plus YO-PRO-1 dye (5 μM). Fluorescence intensity (ex/em = 491/509 nm) is measured after 30 minutes to assess pore formation, and IC50 values are calculated [1] 2. IL-1β release assay: Human PBMCs or rat primary macrophages are primed with LPS (1 μg/mL) for 4 hours. Lu AF27139 (0.1 nM-10 μM) is added 30 minutes before stimulation with ATP (5 mM). Culture supernatants are collected after 1 hour, and IL-1β concentrations are quantified by immunoassay to determine inhibition efficiency [1] 3. Selectivity screening assay: Cells expressing various ion channels (TRPV1, Nav1.7) or GPCRs (CB1, μ-opioid receptor) are used. Lu AF27139 (10 μM) is applied, and functional activity (e.g., channel current, ligand binding) is measured to evaluate off-target effects [1] |
| Animal Protocol |
Animal/Disease Models: Male SD (SD (Sprague-Dawley)) rat (280-350 g); Male C57BL mouse (18-25g) [1] Doses: 3, 10 and 100 mg/kg Route of Administration: Oral Experimental Results: Rat frontal cortex LPS-triggered and BzATP-triggered intracerebroventricular (icv) release were diminished in mice. 1. Pharmacokinetic (PK) study in rats and mice: Rats (n=6 per group) and mice (n=6 per group) are administered Lu AF27139 via oral gavage (1, 3, 10 mg/kg) or intravenous injection (1 mg/kg). Blood samples are collected at 0.25, 0.5, 1, 2, 4, 8, 12, 24 hours post-dose. For CNS distribution analysis, animals are euthanized at 1 hour post-oral dose (3 mg/kg), and brain tissues are harvested. Drug concentrations in plasma and brain homogenates are quantified by LC-MS/MS [1] 2. Murine peritoneal inflammation model: Mice (n=8 per group) are intraperitoneally injected with LPS (5 mg/kg) to prime inflammation. After 4 hours, Lu AF27139 (1, 3, 10 mg/kg) is administered orally. Thirty minutes later, ATP (50 mg/kg) is injected intraperitoneally. Mice are euthanized 1 hour after ATP injection, and peritoneal fluid is collected to measure IL-1β levels [1] 3. Rat neuropathic pain model: Spinal nerve ligation is performed on rats to induce mechanical allodynia. Seven days post-surgery, Lu AF27139 (3, 10 mg/kg) is administered orally once daily for 3 days. Paw withdrawal thresholds to mechanical stimuli are measured before surgery, post-surgery (pre-drug), and 1 hour post-drug administration each day [1] |
| ADME/Pharmacokinetics |
1. Absorption: Oral bioavailability is ~45% (rats) and ~52% (mice) after administration of 3 mg/kg [1] 2. Distribution: Volume of distribution (Vd) is ~1.8 L/kg (rats, IV) and ~1.5 L/kg (mice, IV). Brain/plasma concentration ratio is ~0.8 (rats) and ~0.7 (mice) at 1 hour post-oral 3 mg/kg [1] 3. Metabolism: Minimally metabolized; >85% of the drug is detected as parent compound in rat and mouse plasma [1] 4. Excretion: Elimination half-life (t1/2) is ~2.5 hours (rats, IV), ~3.1 hours (rats, oral 3 mg/kg), ~2.8 hours (mice, oral 3 mg/kg) [1] 5. Clearance: Systemic clearance is ~5.2 mL/min/kg (rats, IV) and ~4.8 mL/min/kg (mice, IV) [1] |
| Toxicity/Toxicokinetics |
1. Acute toxicity: No mortality or obvious toxic signs (e.g., lethargy, weight loss) in rats after oral administration of Lu AF27139 at 1000 mg/kg [1] 2. Plasma protein binding: ~92% in rat plasma and ~90% in human plasma (measured by equilibrium dialysis at 1 μM) [1] 3. No significant changes in liver function markers (ALT, AST) or kidney function markers (BUN, Scr) in rats after 7 days of oral administration at 30 mg/kg/day [1] |
| References |
[1]. Synthesis and Characterization of the Novel Rodent-Active and CNS-Penetrant P2X7 Receptor Antagonist Lu AF27139. J Med Chem. 2021 Apr 22;64(8):4891-4902. [2]. Synthesis and Characterization of the Novel Rodent-Active and CNS-Penetrant P2X7 Receptor Antagonist Lu AF27139. J Med Chem. 2021;64(8):4891-4902. |
| Additional Infomation |
1. Lu AF27139 is a novel, potent, and selective P2X7 receptor antagonist with optimized CNS penetration and pharmacokinetic properties [1] 2. Its chemical structure features a pyrazolo[1,5-a]pyrimidine scaffold, which contributes to high affinity for P2X7 receptor and favorable lipophilicity for BBB crossing [1] 3. Designed for preclinical research to investigate the role of P2X7 receptor in CNS disorders (e.g., neuroinflammation, neuropathic pain, neurodegenerative diseases) [1] 4. Exerts in vivo efficacy by inhibiting P2X7-mediated pro-inflammatory cytokine (IL-1β) release and modulating pain signaling pathways [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~125 mg/mL (~251.04 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (4.18 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (4.18 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (4.18 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0084 mL | 10.0418 mL | 20.0835 mL | |
| 5 mM | 0.4017 mL | 2.0084 mL | 4.0167 mL | |
| 10 mM | 0.2008 mL | 1.0042 mL | 2.0084 mL |