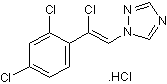
Loreclezole HCl (R72063; R72063; R 72063) is the hydrochloride salt of Loreclezole which is a triazole derivative and an anti-seizure, anticonvulsant and antiepileptic agent. Loreclezole acts as a subtype-selective positive allosteric modulator (PAM) of GABAA receptor.
Physicochemical Properties
| Molecular Formula | C10H7CL4N3 |
| Molecular Weight | 310.994677782059 |
| Exact Mass | 310.936 |
| CAS # | 2227372-56-5 |
| Related CAS # | 117857-45-1; 2227372-56-5 (HCl) |
| PubChem CID | 45073437 |
| Appearance | Typically exists as solid at room temperature |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 2 |
| Rotatable Bond Count | 2 |
| Heavy Atom Count | 17 |
| Complexity | 272 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | C1=CC(=C(C=C1Cl)Cl)/C(=C/N2C=NC=N2)/Cl.Cl |
| InChi Key | AWKSVFLKVCVVFQ-MDZFRNKHSA-N |
| InChi Code | InChI=1S/C10H6Cl3N3.ClH/c11-7-1-2-8(9(12)3-7)10(13)4-16-6-14-5-15-16;/h1-6H;1H/b10-4-; |
| Chemical Name | 1-[(Z)-2-chloro-2-(2,4-dichlorophenyl)ethenyl]-1,2,4-triazole;hydrochloride |
| Synonyms | R72063; R72063; LORECLEZOLE HYDROCHLORIDE; 2227372-56-5; Loreclezole (hydrochloride); DTXSID0045676; (Z)-1-(2-Chloro-2-(2,4-dichlorophenyl)vinyl)-1H-1,2,4-triazole hydrochloride; DTXCID8025676; 1-[(Z)-2-chloro-2-(2,4-dichlorophenyl)ethenyl]-1,2,4-triazole;hydrochloride; NCGC00025098-01; R 72063 HCl |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | GABAA receptor |
| ln Vitro | Loreclezole, an anticonvulsant and antiepileptic compound, potentiates gamma-aminobutyric acid (GABA) type A receptor function, by interacting with a specific allosteric modulatory site on receptor beta-subunits. A similar selectivity for GABAA receptor beta-subunits is apparent for the direct activation of receptor-operated Cl- channels, by the general anesthetics propofol and pentobarbital. The ability of loreclezole to activate GABAA receptors directly has now been compared, biochemically and electrophysiologically, with that of propofol. In well-washed rat cortical membranes (devoid of endogenous GABA), loreclezole and propofol increased t-[35S]butylbicyclophosphorothionate ([35S]TBPS) binding by up to 28% (at 5 microM) and 80% (at 10 microM), respectively. Higher concentrations (50-100 microM) of both compounds inhibited [35S]TBPS binding with great efficacy, an effect mimicked by GABA. In contrast, the benzodiazepine diazepam increased [35S]TBPS binding, but failed to inhibit this parameter, even at high concentrations. At concentrations of 50-100 microM, loreclezole induced inward Cl- currents in the absence of GABA, in Xenopus oocytes expressing human recombinant GABAA receptors, comprised of alpha 1-, beta 2- and gamma 2S-subunits. At 100 microM, the current evoked by loreclezole was 26% of that induced by 5 microM GABA. The current evoked by 100 microM propofol was 98% of that induced by 5 microM GABA. Currents induced by loreclezole, like those evoked by propofol, were potentiated by diazepam in a flumazenil-sensitive manner and blocked by either bicuculline or picrotoxin. These data suggest that loreclezole shares, with propofol, an agonistic action at GABAA receptors containing the beta 2-subunit and that the different efficacies of the two compounds in this regard, may underlie the difference in their pharmacological profiles. The failure of loreclezole to activate GABAA receptors containing the beta 1-subunit may be responsible for its lack of hypnotic effect[2]. |
| ln Vivo | The dose of pentylenetetrazole necessary to trigger convulsion 60 minutes after injection (loreclezole 10, 25, 50, or 75 mg/kg) increased the epileptic threshold in a dose-dependent manner 60 minutes prior to the epileptic threshold test. According to the results of the "Pull-Up" test, loreclezole likewise has little effects on muscular tone loss [3]. |
| Enzyme Assay | Type A gamma-aminobutyric acid (GABAA) receptors of the mammalian nervous system are a family of ligand-gated ion channels probably formed from the coassembly of different subunits (alpha 1-6, beta 1-3, gamma 1-3, delta) in the arrangement alpha beta gamma or alpha beta delta. The activation of these receptors by GABA can be modulated by a range of compounds acting at distinct allosteric sites. One such compound is the broad-spectrum anticonvulsant loreclezole, which we have recently shown to act via a specific modulatory site on the beta subunit of the GABAA receptor. The action of loreclezole depends on the type of beta subunit present in the receptor complex; receptors containing beta 2 or beta 3 subunits have > 300-fold higher affinity for loreclezole than receptors containing a beta 1 subunit. We have used this property to identify the amino acid residue in the beta subunit that determines the subunit selectivity of loreclezole. Chimeric beta 1/beta 2 human GABAA receptor subunits were constructed and coexpressed in Xenopus oocytes with human alpha 1 and gamma 2s subunits. The chimera beta 1/beta 2Lys237-Gly334 conferred sensitivity to 1 microM loreclezole. Within this region there are four amino acids that are conserved in beta 2 and beta 3 but differ in beta 1. By mutating single amino acids of the beta 1 subunit to the beta 2/beta 3 equivalent, only the beta 1 mutation of Ser-290-->Asn conferred potentiation by loreclezole. Similarly, mutation of the homologous residue in the beta 2 and beta 3 subunits to the beta 1 equivalent (Asn-->Ser) resulted in loss of sensitivity to loreclezole. The affinity for GABA and the potentiation by flunitrazepam were unchanged in receptors containing the mutated beta subunits. Thus, a single amino acid, beta 2 Asn-289 (beta 3 Asn-290), located at the carboxyl-terminal end of the putative channel-lining domain TM2, confers sensitivity to the modulatory effects of loreclezole[1]. |
| Animal Protocol |
Animal/Disease Models: Adult male Lister Hooded rat[3]. Doses: 10, 25, 50 or 75 mg/kg. Route of Administration: IP, 60 minutes before measuring seizure threshold. Experimental Results: Produced a dose-dependent increase in epileptic threshold, as measured by the dose of pentylenetetrazole required to produce convulsion after 60 minutes. Loreclezole is an anticonvulsant and anxiolytic compound which has been reported to potentiate GABA via a novel allosteric site on the beta-subunit of the receptor. We have now studied in rats both the in vivo and in vitro pharmacology of the compound. The dose of loreclezole required to increase by 50% the dose of intravenous pentylenetetrazol eliciting a seizure was comparable to that of barbiturates and chlormethiazole (in mg/kg): diazepam, 1.3; pentobarbitone, 16; chlormethiazole, 22; loreclezole, 25; pentobarbitone, 36. Loreclezole dose-dependently decreased locomotion (dose to decrease locomotion by 50% (in mg/kg): chlormethiazole, 9; pentobarbitone, 16; loreclezole, 25). Loreclezole, chlormethiazole and pentobarbitone all failed to displace [3H]muscimol and [3H]flunitrazepam binding from a rat cortical membrane preparation. All three compounds fully displaced [35S]TBPS binding (IC50 values: loreclezole, 4.34 +/- 0.68 microM; pentobarbitone, 37.39 +/- 3.24 microM; chlormethiazole, 82.10 +/- 8.52 microM). Addition of bicuculline (10 microM) produced a major rightward shift in the loreclezole and pentobarbitone displacement curves, increasing IC50 values for [35S]TBPS binding by 25 times (loreclezole), 6 times (pentobarbitone) and 2.7 times (chlormethiazole), suggesting a greater involvement of GABA in the interaction of loreclezole with the chloride channel than in the case of chlormethiazole. Anticonvulsant activity of the compounds did not appear to relate to [35S]TBPS binding activity. Other binding data suggested that although the evidence of others indicates that loreclezole interacts with a specific allosteric site on the beta-subunit, it nevertheless also alters the binding characteristics of other modulatory sites.[3] |
| References |
[1]. Wingrove PB, et al. The modulatory action of loreclezole at the gamma-aminobutyric acid type A receptor is determined by a single amino acid in the beta 2 and beta 3 subunit. Proc Natl Acad Sci U S A. 1994 May 10;91(10):4569-73. [2]. Sanna E, et al. Direct activation of GABAA receptors by loreclezole, an anticonvulsant drug with selectivity for the beta-subunit. Neuropharmacology. 1996;35(12):1753-60. [3]. Green AR, et al. A behavioural and neurochemical study in rats of the pharmacology of loreclezole, a novel allosteric modulator of the GABAA receptor. Neuropharmacology. 1996;35(9-10):1243-50. |
| Additional Infomation | Cerebellar Purkinje cells (PCs) are particularly sensitive to cerebral ischemia, and decreased GABA(A) receptor function following injury is thought to contribute to PC sensitivity to ischemia-induced excitotoxicity. Here we examined the functional properties of the GABA(A) receptors that are spared following ischemia in cultured Purkinje cells from rat and in vivo ischemia in mouse. Using subunit-specific positive modulators of GABA(A) receptors, we observed that oxygen and glucose deprivation (OGD) and cardiac arrest-induced cerebral ischemia cause a decrease in sensitivity to the β(2/3) -subunit-preferring compound, etomidate. However, sensitivity to propofol, a β-subunit-acting compound that modulates β(1-3) -subunits, was not affected by OGD. The α/γ-subunit-acting compounds, diazepam and zolpidem, were also unaffected by OGD. We performed single-cell reverse transcription-polymerase chain reaction on isolated PCs from acutely dissociated cerebellar tissue and observed that PCs expressed the β(1) -subunit, contrary to previous reports examining GABA(A) receptor subunit expression in PCs. GABA(A) receptor β(1) -subunit protein was also detected in cultured PCs by western blot and by immunohistochemistry in the adult mouse cerebellum and levels remained unaffected by ischemia. High concentrations of loreclezole (30 μm) inhibited PC GABA-mediated currents, as previously demonstrated with β(1) -subunit-containing GABA(A) receptors expressed in heterologous systems. From our data we conclude that PCs express the β(1) -subunit and that there is a greater contribution of β(1) -subunit-containing GABA(A) receptors following OGD.https://pubmed.ncbi.nlm.nih.gov/23176253/ |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2155 mL | 16.0777 mL | 32.1554 mL | |
| 5 mM | 0.6431 mL | 3.2155 mL | 6.4311 mL | |
| 10 mM | 0.3216 mL | 1.6078 mL | 3.2155 mL |