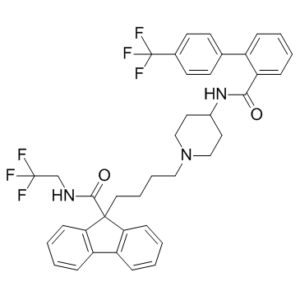
Lomitapide (formerly known as AEGR-733; BMS-201038; Juxtapid; Lojuxta) is a novel, oral and potent inhibitor of microsomal triglyceride-transfer protein (MTP) approved as a lipid-lowering agent for the treatment of homozygous familial hypercholesterolemia. It inhibits MTP with an IC50 of 8 nM in vitro. As an orally active MTP inhibitor, Lomitapide is indicated as an adjunct to a low-fat diet and other lipid-lowering treatments, including LDL apheresis where available for the reduction of LDL-C, total cholesterol, apolipoprotein B, and non-high-density lipoprotein cholesterol in adult patients with HoFH.
Physicochemical Properties
| Molecular Formula | C39H37N3O2F6 |
| Molecular Weight | 693.72038 |
| Exact Mass | 693.278 |
| CAS # | 182431-12-5 |
| Related CAS # | Lomitapide mesylate;202914-84-9;Lomitapide-d8;2459377-96-7 |
| PubChem CID | 9853053 |
| Appearance | White to off-white solid powder |
| Density | 1.3±0.1 g/cm3 |
| Boiling Point | 778.2±60.0 °C at 760 mmHg |
| Flash Point | 424.4±32.9 °C |
| Vapour Pressure | 0.0±2.7 mmHg at 25°C |
| Index of Refraction | 1.606 |
| LogP | 7.78 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 9 |
| Rotatable Bond Count | 10 |
| Heavy Atom Count | 50 |
| Complexity | 1110 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | MBBCVAKAJPKAKM-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C39H37F6N3O2/c40-38(41,42)25-46-36(50)37(33-13-5-3-10-30(33)31-11-4-6-14-34(31)37)21-7-8-22-48-23-19-28(20-24-48)47-35(49)32-12-2-1-9-29(32)26-15-17-27(18-16-26)39(43,44)45/h1-6,9-18,28H,7-8,19-25H2,(H,46,50)(H,47,49) |
| Chemical Name | N-(2,2,2-trifluoroethyl)-9-(4-(4-(4'-(trifluoromethyl)-[1,1'-biphenyl]-2-carboxamido)piperidin-1-yl)butyl)-9H-fluorene-9-carboxamide |
| Synonyms | BMS 201038; AEGR733; BMS-201038; AEGR-733; BMS201038; BMS 201038-01; AEGR 733; Lomitapide mesylate. trade name: Juxtapid; Lojuxta. |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
|
|
| ln Vitro |
An oral microsomal triglyceride transfer protein (MTP) inhibitor called lipitpide is used to treat patients with homoeopathy of function (HoFH), an uncommon type of hypercholesterolemia that causes early atherosclerotic disease. Cytochrome P-450 (CYP) isoenzyme 3A4 is responsible for the hepatic metabolism of lomitapide. It has an interaction with CYP3A4 substrates, such as Simvastatin and Atorvastatin [2]. 1. Lomitapide potently and selectively inhibited triglyceride transfer activity of human hepatic MTP in a concentration-dependent manner, with an IC50 of 0.19 μM; it showed no significant inhibition of other lipid transfer proteins (e.g., phospholipid transfer protein, IC50 > 100 μM) [2] 2. In human hepatocellular carcinoma HepG2 cells, Lomitapide (0.1–10 μM) concentration-dependently suppressed the secretion of apolipoprotein B (ApoB) (IC50 = 0.5 μM) and inhibited the assembly and secretion of very-low-density lipoprotein (VLDL) particles, as detected by ELISA and Western blot analysis of cell supernatants [2] |
|
| ln Vivo |
Lomitapide lowers plasma concentrations of low-density lipoprotein cholesterol (LDL-C) by an average of more than 50% when used by itself or in conjunction with other lipid-lowering medications. Significant gastrointestinal side effects and elevated levels of hepatic fat are linked to lomitapide. For 50 mg of lomitapide, the bioavailability is 7.1%. Lomapide has an average half-life of 39.7 hours [2]. It was demonstrated that lomitapide, at doses of 0.3 mg/kg and 1 mg/kg, respectively, reduced serum triglycerides by 35% and 47% after just one treatment. Dose-dependent decreases in triglycerides (71%–87%), nonesterified fatty acids (33%–40%), and low-density lipoprotein cholesterol (26–29%) were also observed after multiple-dose lomitapide treatment [3]. 1. In patients with homozygous familial hypercholesterolemia (HoFH), oral administration of Lomitapide (starting dose 5 mg/day, titrated up to a maximum of 60 mg/day) resulted in a mean reduction of 50% in low-density lipoprotein cholesterol (LDL-C) levels, along with significant decreases in total cholesterol (38%), triglycerides (45%), and apolipoprotein B (47%) after 24 weeks of treatment [2] 2. In Zucker fatty rats (a model of obesity and insulin resistance), oral Lomitapide (10 mg/kg/day for 4 weeks) reduced serum triglyceride levels by 40%, LDL-C by 35%, and increased insulin sensitivity: fasting insulin levels decreased by 25%, and glucose tolerance was significantly improved in the oral glucose tolerance test (OGTT) [3] 3. Lomitapide (10 mg/kg/day, p.o. for 4 weeks) reduced the area of atherosclerotic plaques in the aorta of Zucker fatty rats by 30% and decreased hepatic triglyceride content by 22% compared with vehicle-treated controls [3] |
|
| Enzyme Assay |
1. Human hepatic MTP activity assay [2]: Human liver microsomes were isolated as the source of MTP and incubated with lipid vesicles composed of [3H]-triglyceride, phosphatidylcholine, and cholesterol in the presence of serial concentrations of Lomitapide. The reaction was incubated at 37°C for 30 minutes, then lipid vesicles and microsomes were separated by ultracentrifugation. The radioactivity of [3H]-triglyceride transferred to microsomes was measured by liquid scintillation counting to calculate the MTP transfer activity inhibition rate and determine the IC50 value. 2. Lipid transfer protein selectivity assay [2]: Lomitapide was tested at concentrations up to 100 μM against phospholipid transfer protein (PLTP) and cholesterol ester transfer protein (CETP) using the same radiolabeled lipid transfer assay as for MTP; the percentage of enzyme inhibition was calculated to evaluate selectivity. |
|
| Cell Assay |
1. ApoB secretion inhibition assay in HepG2 cells [2]: HepG2 cells were seeded in 24-well plates at a density of 5×10⁵ cells/well and cultured in serum-free medium overnight. The cells were treated with Lomitapide (0.1–10 μM) for 24 hours, and the culture supernatant was collected. ApoB levels in the supernatant were quantified by ELISA, and VLDL secretion was assessed by measuring triglyceride content in the d < 1.006 g/mL fraction of the supernatant. For Western blot analysis, cell lysates and supernatants were subjected to SDS-PAGE, probed with anti-ApoB antibody, and band intensity was quantified by densitometry. 2. Cell viability assay (MTT) [2]: HepG2 cells were seeded in 96-well plates and treated with Lomitapide (0.01–100 μM) for 72 hours. MTT solution was added, and after 4 hours of incubation, formazan crystals were dissolved with dimethyl sulfoxide. Absorbance at 570 nm was measured to calculate cell viability, and the CC50 (50% cytotoxic concentration) was determined to be >100 μM. |
|
| Animal Protocol |
1. Zucker fatty rat model of obesity and insulin resistance [3]: Male Zucker fatty rats (6 weeks old) were randomly divided into vehicle and Lomitapide-treated groups (n=8 per group). Lomitapide was formulated in 0.5% methylcellulose in water and administered orally by gavage at a dose of 10 mg/kg once daily for 4 weeks. Body weight and fasting blood glucose levels were measured weekly. At the end of the treatment period, blood samples were collected to determine serum lipid profiles (triglycerides, LDL-C, HDL-C, total cholesterol) and insulin levels. Liver tissues were excised to measure hepatic triglyceride content, and aortic tissues were analyzed for atherosclerotic plaque area by oil red O staining. 2. Preclinical animal studies for HoFH [2]: In C57BL/6 mice and cynomolgus monkeys, Lomitapide was administered orally at doses of 5–20 mg/kg/day (mice) and 1–5 mg/kg/day (monkeys) for 4–8 weeks. Plasma lipid levels (LDL-C, triglycerides, ApoB) were measured at weekly intervals, and liver histology was examined to assess lipid accumulation. |
|
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion In healthy patients, time to maximum lomitapide concentration is about 6 hours with a single dose of 60 mg. Lomitapide has an approximate absolute bioavailability of 7%. About 52.9-59.5% is eliminated by the urine and 33.4-35.1% is eliminated by the feces. The steady state volume of distribution is about 985-1292 L. The mean lomitapide volume of distribution at steady state is 985-1292 liters. Lomitapide is 99.8% plasma-protein bound. Upon oral administration of a single 60-mg dose of Juxtapid, the lomitapide tmax is around 6 hours in healthy volunteers. The absolute bioavailability of lomitapide is approximately 7%. Lomitapide pharmacokinetics is approximately dose-proportional for oral single doses from 10-100 mg. In a mass-balance study, a mean of 59.5% and 33.4% of the dose was excreted in the urine and feces, respectively. In another mass-balance study, a mean of 52.9% and 35.1% of the dose was excreted in the urine and feces, respectively. Lomitapide was not detectable in urine samples. M1 is the major urinary metabolite. Lomitapide is the major component in the feces. It is not known whether lomitapide is excreted in human milk. Metabolism / Metabolites Lomitapide is mainly metabolized by CYP3A4 to it's inactive metabolites, M1 and M3. CYP enzymes that metabolize lomitapide to a minor extent include CYP 1A2,2B6,2C8,2C19. Lomitapide is metabolized extensively by the liver. The metabolic pathways include oxidation, oxidative N-dealkylation, glucuronide conjugation, and piperidine ring opening. Cytochrome P450 (CYP) 3A4 metabolizes lomitapide to its major metabolites, M1 and M3, as detected in plasma. The oxidative N-dealkylation pathway breaks the lomitapide molecule into M1 and M3. M1 is the moiety that retains the piperidine ring, whereas M3 retains the rest of the lomitapide molecule in vitro. CYPs 1A2, 2B6, 2C8, and 2C19 may metabolize lomitapide to a small extent to M1. M1 and M3 do not inhibit activity of microsomal triglyceride transfer protein in vitro. Biological Half-Life Lomitapide half-life is about 39.7 hours. The mean lomitapide terminal half-life is 39.7 hours. 1. Absorption: Lomitapide has low oral bioavailability (approximately 7%) in humans due to extensive first-pass metabolism in the liver; peak plasma concentrations (Cmax) are reached at 4–6 hours after oral administration [2] 2. Distribution: Lomitapide has a high volume of distribution (Vd) of approximately 94 L in humans and exhibits extensive tissue distribution, with the highest concentrations in the liver (the primary target organ) [2] 3. Metabolism: Lomitapide is primarily metabolized in the liver by cytochrome P450 3A4 (CYP3A4); major metabolites include hydroxylated and demethylated derivatives, which are inactive against MTP [2] 4. Elimination: The terminal half-life (t1/2) of Lomitapide in humans is approximately 40 hours; approximately 82% of the administered dose is excreted in feces (mostly as metabolites), and <1% is excreted in urine [2] 5. Plasma protein binding: Lomitapide has a plasma protein binding rate of >99% in human plasma [2] |
|
| Toxicity/Toxicokinetics |
Toxicity Summary IDENTIFICATION AND USE: Lomitapide is a white to off-white powder. Lomitapide is indicated as an adjunct to a low-fat diet and other lipid-lowering treatments, including LDL apheresis where available, to reduce low-density lipoprotein cholesterol (LDL-C), total cholesterol (TC), apolipoprotein B (apo B), and non-high-density lipoprotein cholesterol (non-HDL-C) in patients with homozygous familial hypercholesterolemia (HoFH). HUMAN EXPOSURE AND TOXICITY: Very few data are available on the effects of overdose to lomitapide. The maximum dose administered to human subjects in clinical studies was 200 mg lomitapide, as a single dose, without adverse consequences. Lomitapide should not be used during pregnancy because lomitapide may cause fetal harm when administered to a pregnant woman. Lomitapide may also cause diarrhea and malabsorption in patients with rare hereditary disorders, including galactose intolerance, the Lapp lactase deficiency, and glucose-galactose malabsorption; therefore, use of lomitapide should be avoided in such patients. Lomitapide causes a risk of hepatotoxicity. Lomitapide can cause elevations in transaminases and hepatic steatosis. To what extent lomitapide-associated hepatic steatosis promotes the elevations in transaminases is unknown. Although cases of hepatic dysfunction or hepatic failure have not been reported, there is concern that lomitapide could induce steatohepatitis, which can progress to cirrhosis over several years. Therefore, it should not be administered to patients with moderate or severe hepatic impairment (Child-Pugh class B or C) or patients with active liver disease, including unexplained, persistent elevations in serum aminotransferase concentrations. Lomitapide did not exhibit genotoxic potential in a battery of studies, including an in vitro cytogenetics assay using primary human lymphocytes. ANIMAL STUDIES: In a 2-year dietary carcinogenicity study in mice, lomitapide was administered at doses of 0.3, 1.5, 7.5, 15, or 45 mg/kg/day. There were statistically significant increases in the incidences of liver adenomas and carcinomas in males at doses as low as 1.5 mg/kg/day and in females at as low as 7.5 mg/kg/day. Incidences of small intestinal carcinomas in males and combined adenomas and carcinomas in females were significantly increased at doses as low as 15 mg/kg/day. In 2 year studies in rats, there were no statistically significant drug-related increases in tumor incidences. Reproduction studies were conducted in rats, rabbits and ferrets. Oral gavage doses of 0.04, 0.4, or 4 mg/kg/day lomitapide given to pregnant rats from gestation day 6 through organogenesis were associated with fetal malformations at greater than or equal to 2-times human exposure at the maximum recommended human dose (MRHD) (60 mg) based on plasma AUC comparisons. Fetal malformations included umbilical hernia, gastroschisis, imperforate anus, alterations in heart shape and size, limb malrotations, skeletal malformations of the tail, and delayed ossification of cranial, vertebral and pelvic bones. Oral gavage doses of 1.6, 4, 10, or 25 mg/kg/day lomitapide given to pregnant ferrets from gestation day 12 through organogenesis were associated with both maternal toxicity and fetal malformations at exposures that ranged from less than the human exposure at the MRHD to 5-times the human exposure at the MRHD. Fetal malformations included umbilical hernia, medially rotated or short limbs, absent or fused digits on paws, cleft palate, open eye lids, low-set ears, and kinked tail. In rabbits, exposures up to 3 times the MRHD based on body surface area (BSA) (MRDH-BSA) from gestational day 6 through organogenesis were not associated with adverse effects. However, exposure equal to or greater than 6 times the MRHD-BSA resulted in embryo-fetal death. Lomitapide had no effect on fertility in rats at doses up to 5 mg/kg/day at systemic exposures estimated to be 4-times (females) and 5-times (males) higher than in humans at 60 mg based on AUC. Lomitapide did not exhibit genotoxic potential in a battery of studies, including the in vitro Bacterial Reverse Mutation (Ames) and an oral micronucleus study in rats. Hepatotoxicity Lomitapide is associated with a moderately high rate of serum aminotransferase elevations during therapy, levels above 3 times the upper limit of normal (ULN) occurring in 34% of patients. Aminotransferase elevations above 10 times ULN have also been reported which can necessitate drug discontinuation. Despite the frequency of ALT elevations, however, increases in serum bilirubin and alkaline phosphatase levels are rare and there have been no reports of clinically apparent acute liver injury with jaundice. Chronic therapy with lomitapide can be associated with fluctuations in serum aminotransferase levels and accumulation of liver fat. In some instances, the increase in liver fat is from baseline levels of Likelihood score: C (probable cause of clinically significant liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation No relevant published information exists with the use of lomitapide during breastfeeding. Because of a concern with disruption of infant lipid metabolism and possible tumorigenicity, lomitapide should not be used during breastfeeding. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Plasma protein binding is about 99.8% Interactions Concomitant use of lomitapide with potent (e.g., boceprevir, clarithromycin, conivaptan, indinavir, itraconazole, ketoconazole, lopinavir/ritonavir, nefazodone, nelfinavir, posaconazole, ritonavir, saquinavir, telaprevir, telithromycin, tipranavir/ritonavir, voriconazole) or moderate CYP3A4 inhibitors (e.g., aprepitant, atazanavir, ciprofloxacin, crizotinib, darunavir/ritonavir, diltiazem, erythromycin, fluconazole, fosamprenavir, imatinib, verapamil) is contraindicated. If concomitant use with moderate or potent CYP3A4 inhibitors cannot be avoided, lomitapide therapy should be interrupted during the course of treatment with the CYP3A4 inhibitor. Concomitant use of lomitapide with inhibitors of CYP3A4 may result in increased systemic exposure to lomitapide. When the potent CYP3A4 inhibitor ketoconazole (200 mg twice daily for 9 days) was administered concomitantly with lomitapide (60 mg once daily), peak plasma concentration and area under the plasma concentration-time curve (AUC) of lomitapide were increased by 15- and 27-fold, respectively. When the potent CYP3A4 inhibitor clarithromycin was added to lomitapide therapy in at least 1 patient, ALT and AST concentrations were increased to 24 and 13 times the upper limit of normal (ULN), respectively, within days of initiating the potent CYP3A4 inhibitor. Concomitant use of lomitapide with moderate inhibitors of CYP3A4 has not been studied; however, results of pharmacokinetic studies evaluating concomitant use of lomitapide with potent and weak CYP3A4 inhibitors suggest that moderate CYP3A4 inhibitors will likely increase lomitapide exposure. Following concomitant use of lomitapide (10 mg once daily for 7 days) with fenofibrate (single 145-mg dose as micronized formulation), peak plasma concentration and AUC of fenofibric acid were decreased by 29 and 10%, respectively. Dosage adjustment of fenofibrate (as micronized formulation) is not required during concomitant use with lomitapide. Following concomitant use of lomitapide (10 mg once daily for 7 days) with ezetimibe (single 10-mg dose), peak plasma concentration and AUC of ezetimibe were increased by 3 and 6%, respectively. Dosage adjustment of ezetimibe is not required during concomitant use with lomitapide. For more Interactions (Complete) data for Lomitapide (18 total), please visit the HSDB record page. 1. In vitro cytotoxicity: Lomitapide showed low cytotoxicity in human HepG2 cells, with a CC50 > 100 μM [2] 2. Clinical adverse effects: The most common adverse effect of Lomitapide in HoFH patients is gastrointestinal symptoms (nausea, diarrhea, vomiting), occurring in approximately 80% of patients; these symptoms are mild to moderate and usually resolve with dose titration [2] 3. Hepatic toxicity: Long-term use of Lomitapide may cause mild hepatic steatosis (increased liver fat content by ~15% in clinical trials), but no significant elevations in liver transaminases (ALT/AST) or liver dysfunction were observed [2] 4. Drug-drug interaction: Lomitapide is a substrate of CYP3A4; strong CYP3A4 inhibitors (e.g., ketoconazole) increase its plasma concentration by approximately 20-fold, and strong CYP3A4 inducers (e.g., rifampicin) decrease its concentration by >50%. Co-administration with strong CYP3A4 inhibitors is contraindicated [2] 5. Animal toxicity: In Zucker fatty rats treated with Lomitapide (10 mg/kg/day for 4 weeks), no significant weight loss, renal toxicity, or cardiovascular toxicity was observed; mild hepatic lipid accumulation (15% increase in liver triglyceride content) was noted but reversible upon drug discontinuation [3] |
|
| References |
[1]. 5-Carboxamido-1,3,2-dioxaphosphorinanes, potent inhibitors of MTP. Bioorg Med Chem Lett. 2004 Oct 18;14(20):5067-70. [2]. Lomitapide: A novel agent for the treatment of homozygous familial hypercholesterolemia. Am J Health Syst Pharm. 2014 Jun 15;71(12):1001-8. [3]. Inhibition of microsomal triglyceride transfer protein improves insulin sensitivity and reduces atherogenic risk in Zucker fatty rats. Clin Exp Pharmacol Physiol. 2011 May;38(5):338-44. |
|
| Additional Infomation |
Lomitapide is a member of the class of benzamides obtained by formal condensation of the carboxy group of 4'-(trifluoromethyl)biphenyl-2-carboxylic acid with the primary amino group of 9-[4-(4-aminopiperidin-1-yl)butyl]-N-(2,2,2-trifluoroethyl)-9H-fluorene-9-carboxamide. Used (as its mesylate salt) as a complement to a low-fat diet and other lipid-lowering treatments in patients with homozygous familial hypercholesterolemia. It has a role as an anticholesteremic drug and a MTP inhibitor. It is a member of piperidines, a member of fluorenes, a member of benzamides and a member of (trifluoromethyl)benzenes. It is a conjugate base of a lomitapide(1+). Lomitapide is a microsomal triglyceride transfer protein (MTP) inhibitor used in homozygous familial hypercholesterolemia (HoFH) patients. It is marketed under the name Juxtapid (R). Lomitapide is a Microsomal Triglyceride Transfer Protein Inhibitor. The mechanism of action of lomitapide is as a Microsomal Triglyceride Transfer Protein Inhibitor, and Cytochrome P450 3A4 Inhibitor, and P-Glycoprotein Inhibitor. Lomitapide is a cholesterol lowering agent that acts by inhibition of the microsomal triglyceride transfer protein and is used to treat the severe lipid abnormalities of familial hypercholesterolemia. Lomitapide is associated with mild, asymptomatic and self-limited serum aminotransferase elevations during therapy that are usually accompanied by an increase in hepatic fat. Long term therapy with lomitapide has been linked to development of steatohepatitis and hepatic fibrosis. Lomitapide is a small molecule inhibitor of microsomal triglyceride transfer protein (MTP), an enzyme located in the lumen of the endoplasmic reticulum responsible for absorbing dietary lipids and transferring triglycerides onto apolipoprotein B (apo-B) in the assembly of very-low-density lipoprotein. Inhibition of MTP by lomitapide blocks transfer of lipid to apo-B, and as a result emerging apo-B is destroyed and lipoprotein secretion is inhibited. See also: Lomitapide Mesylate (has salt form). Drug Indication Used in homozygous familial hypercholesterolemia (HoFH) patients to reduce low-density lipoprotein cholesterol (LDL-C), total cholesterol (TC), apolipoprotein B (apo B), and non-high-density lipoprotein cholesterol (non-HDL-C). FDA Label Lojuxta is indicated as an adjunct to a lowâfat diet and other lipidâlowering medicinal products with or without low-density-lipoprotein (LDL) apheresis in adult patients with homozygous familial hypercholesterolaemia (HoFH). , , Genetic confirmation of HoFH should be obtained whenever possible. Other forms of primary hyperlipoproteinaemia and secondary causes of hypercholesterolaemia (e. g. nephrotic syndrome, hypothyroidism) must be excluded. , Treatment of (heterozygous or homozygous) familial hypercholesterolaemia Mechanism of Action Within the lumen of the endoplasmic reticulum, lomitapide inhibits microsomal triglyceride transfer protein (MTP), which prevents the formation of apolipoprotein B, and, thus, the formation of VLDL and chylomicrons as well. Altogether, this leads to a reduction of low-density lipoprotein cholesterol. Juxtapid directly binds and inhibits microsomal triglyceride transfer protein (MTP), which resides in the lumen of the endoplasmic reticulum, thereby preventing the assembly of apo B-containing lipoproteins in enterocytes and hepatocytes. This inhibits the synthesis of chylomicrons and very low-density lipoprotein (VLDL). The inhibition of the synthesis of VLDL leads to reduced levels of plasma low-density lipoprotein cholesterol (LDL-C). Therapeutic Uses Juxtapid is indicated as an adjunct to a low-fat diet and other lipid-lowering treatments, including LDL apheresis where available, to reduce low-density lipoprotein cholesterol (LDL-C), total cholesterol (TC), apolipoprotein B (apo B), and non-high-density lipoprotein cholesterol (non-HDL-C) in patients with homozygous familial hypercholesterolemia (HoFH). /Included in US product label/ Because of the risk of hepatotoxicity, lomitapide is available only under a restricted distribution program (Juxtapid Risk Evaluation and Mitigation Strategies (REMS)). Healthcare providers and pharmacies must be certified with the Juxtapid REMS program before they can prescribe or dispense lomitapide. Drug Warnings /BOXED WARNING/ WARNING: RISK OF HEPATOTOXICITY. Juxtapid can cause elevations in transaminases. In the Juxtapid clinical trial, 10 (34%) of the 29 patients treated with Juxtapid had at least one elevation in alanine aminotransferase (ALT) or aspartate aminotransferase (AST) > or =3x upper limit of normal (ULN). There were no concomitant clinically meaningful elevations of total bilirubin, international normalized ratio (INR), or alkaline phosphatase. Juxtapid also increases hepatic fat, with or without concomitant increases in transaminases. The median absolute increase in hepatic fat was 6% after both 26 and 78 weeks of treatment, from 1% at baseline, measured by magnetic resonance spectroscopy. Hepatic steatosis associated with Juxtapid treatment may be a risk factor for progressive liver disease, including steatohepatitis and cirrhosis. Measure ALT, AST, alkaline phosphatase, and total bilirubin before initiating treatment and then ALT and AST regularly as recommended. During treatment, adjust the dose of Juxtapid if the ALT or AST are > or =3x ULN. Discontinue Juxtapid for clinically significant liver toxicity. Because of the risk of hepatotoxicity, Juxtapid is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Juxtapid REMS Program. FDA Pregnancy Risk Category: X /CONTRAINDICATED IN PREGNANCY. Studies in animals or humans, or investigational or post-marketing reports, have demonstrated positive evidence of fetal abnormalities or risk which clearly outweights any possible benefit to the patient./ Juxtapid can cause elevations in transaminases and hepatic steatosis, ... . To what extent Juxtapid-associated hepatic steatosis promotes the elevations in transaminases is unknown. Although cases of hepatic dysfunction (elevated transaminases with increase in bilirubin or international normalized ratio (INR)) or hepatic failure have not been reported, there is concern that Juxtapid could induce steatohepatitis, which can progress to cirrhosis over several years. The clinical studies supporting the safety and efficacy of Juxtapid in homozygous familial hypercholesterolemia (HoFH) would have been unlikely to detect this adverse outcome given their size and duration. Consistent with the mechanism of action of Juxtapid (lomitapide), most treated patients exhibited increases in hepatic triglyceride content, with or without concomitant increases in hepatic transaminases. In an open-label Phase 3 study, 18 of 23 patients with homozygous familial hypercholesterolemia (HoFH) developed hepatic steatosis, i.e., hepatic fat > 5.6%, as measured by nuclear magnetic resonance spectroscopy (NMRS). There was a mean increase in absolute hepatic fat content of 6% after both 26 weeks and 78 weeks of treatment, from a mean of 1% at baseline. Clinical data suggest that hepatic fat accumulation is reversible after stopping treatment with Juxtapid, generally over 4 to 6 weeks, but whether histological sequelae remain is unknown, especially after long-term use. The long term consequences of hepatic steatosis associated with Juxtapid treatment are unknown, including risk of progression to steatohepatitis and cirrhosis. For more Drug Warnings (Complete) data for Lomitapide (22 total), please visit the HSDB record page. Pharmacodynamics Lomitapide directly inhibits microsomal triglyceride transfer protein (MTP). 1. Lomitapide is a first-in-class microsomal triglyceride transfer protein (MTP) inhibitor approved by the FDA in 2012 (trade name: Juxtapid) for the treatment of homozygous familial hypercholesterolemia (HoFH) [2] 2. The mechanism of action of Lomitapide involves inhibiting MTP-mediated transfer of triglycerides into nascent VLDL particles in the liver and intestine, thereby reducing VLDL secretion and plasma LDL-C levels [2] 3. Lomitapide is indicated as an adjunct to a low-fat diet and other lipid-lowering therapies (e.g., statins, LDL apheresis) for the treatment of HoFH in adults and adolescents ≥12 years of age [2] 4. Beyond lipid-lowering effects, Lomitapide improves insulin sensitivity and reduces atherosclerotic plaque formation in Zucker fatty rats, suggesting potential applications in metabolic syndrome and cardiovascular disease [3] 5. Literature [1] focuses on 5-carboxamido-1,3,2-dioxaphosphorinanes as novel MTP inhibitors and does not mention Lomitapide [1] 6. Lomitapide has a narrow therapeutic window and requires close monitoring of liver function and lipid levels during clinical use [2] |
Solubility Data
| Solubility (In Vitro) | DMSO : ≥ 100 mg/mL (~144.15 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (3.60 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (3.60 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (3.60 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.4415 mL | 7.2075 mL | 14.4150 mL | |
| 5 mM | 0.2883 mL | 1.4415 mL | 2.8830 mL | |
| 10 mM | 0.1442 mL | 0.7208 mL | 1.4415 mL |