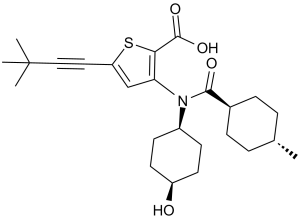
Lomibuvir (also called VX-22; S-1480; VCH222; VX22; S1480; VX222; VCH-222; VX-222) is a novel, potent and selective non-nucleoside inhibitor/NNI that targets the NS5B RNA-dependent RNA polymerase of the hepatitis C virus (HCV), with IC50 values ranging from 0.94 to 1.2 μM. For the treatment of chronic HCV and chronic HCV infections, it has been studied in clinical trials. In patients with genotype 1 HCV infection who were chronically infected, VX-222 showed significant reductions in plasma HCV RNA in phase 1 and 2 clinical trials, indicating effective antiviral efficacy.
Physicochemical Properties
| Molecular Formula | C25H35NO4S |
| Molecular Weight | 445.61 |
| Exact Mass | 445.228 |
| Elemental Analysis | C, 67.38; H, 7.92; N, 3.14; O, 14.36; S, 7.19 |
| CAS # | 1026785-55-6 |
| Related CAS # | Lomibuvir;1026785-55-6; 1026785-59-0 |
| PubChem CID | 24798764 |
| Appearance | White to off-white solid powder |
| Density | 1.2±0.1 g/cm3 |
| Boiling Point | 640.5±55.0 °C at 760 mmHg |
| Flash Point | 341.2±31.5 °C |
| Vapour Pressure | 0.0±2.0 mmHg at 25°C |
| Index of Refraction | 1.589 |
| LogP | 5.15 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 5 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 31 |
| Complexity | 717 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | O=C(C1=C(N([C@H]2CC[C@H](O)CC2)C([C@H]3CC[C@H](C)CC3)=O)C=C(C#CC(C)(C)C)S1)O |
| InChi Key | WPMJNLCLKAKMLA-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C25H35NO4S/c1-16-5-7-17(8-6-16)23(28)26(18-9-11-19(27)12-10-18)21-15-20(13-14-25(2,3)4)31-22(21)24(29)30/h15-19,27H,5-12H2,1-4H3,(H,29,30) |
| Chemical Name | 5-(3,3-dimethylbut-1-ynyl)-3-[(4-hydroxycyclohexyl)-(4-methylcyclohexanecarbonyl)amino]thiophene-2-carboxylic acid |
| Synonyms | TD-4208; TD4208; GSK-1160724; GSK-1160724; Lomibuvir; VX-222; 1026785-59-0; 1026785-55-6; VCH-222; VX-222 (VCH-222, Lomibuvir); cis-Lomibuvir; Lomibuvir (VX-222); TD 4208; GSK1160724; trade name: Yupelri; TD-4208; GSK 1160724 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
HCV NS5B 1a (IC50 = 0.94 μM); HCV NS5B 1b (IC50 = 1.2 μM) Lomibuvir (VX-222) is a potent inhibitor of hepatitis C virus (HCV) NS5B RNA-dependent RNA polymerase (the viral enzyme responsible for viral genome replication), with an IC50 of 16 nM for HCV genotype 1a NS5B polymerase and 12 nM for genotype 1b NS5B polymerase in cell-free enzyme assays [1] - Lomibuvir acts as a non-nucleoside inhibitor (NNI) of HCV NS5B, binding to the thumb domain of the enzyme; it shows no significant inhibition of human RNA polymerases (e.g., Pol I, Pol II) at concentrations up to 10 μM [1] |
| ln Vitro |
VX-222 attaches itself to the HCV RNA-dependent RNA polymerase's thumb II allosteric pocket. With an IC50 of 0.94 and 1.2 μM, respectively, VX-222 shows non-competitive and selective inhibition in HCV NS5B of genotypes 1a and 1b. Subgenomic HCV genotypes 1a and 1b are specifically inhibited by VX-222, with EC50 values of 22.3 and 11.2 nM, respectively. (Source: ) Similarly, VX-222 inhibits the 1b/Con1 HCV subgenomic replicon with an EC50 of 5 nM, according to a recent study. While having little to no effect on de novo-initiated RNA synthesis, VX-222 preferentially inhibits primer-dependent RNA synthesis. In cell-free assays with recombinant HCV genotype 1a NS5B polymerase, 50 nM Lomibuvir inhibited RNA synthesis by ~90% (measured via incorporation of radioactive [³H]-UTP into newly synthesized RNA); this inhibition was reversible upon drug removal [1] - In HCV genotype 1a (H77 strain) replicon cells, treatment with 25 nM Lomibuvir for 48 hours reduced HCV RNA levels by ~95% (qRT-PCR) and decreased viral protein (NS5B) expression by ~90% (Western blot); the EC50 for HCV genotype 1a replicon was 22 nM, and for genotype 1b (Con1 strain) replicon was 18 nM [1] - In combination with filibuvir (another HCV NS5B inhibitor) in HCV 1a replicon cells, 10 nM Lomibuvir + 10 nM filibuvir showed synergistic inhibition, reducing HCV RNA by ~99% (vs. ~70% and ~65% for single drugs at the same concentration) [1] |
| ln Vivo | VCH-222 exhibits fine pharmacokinetic properties in rats and dogs, such as low total body clearance, excellent oral bioavailability (more than 30%), and good ADME characteristics. Multiple enzymes (CYP1A1, 2A6, 2B6, 2C8, CYP 3A4, UGT1A3) biotransform VCH-222. It is expected that VCH-222 is actively transported in the liver and excreted mostly intact in bile or as glucuronide adducts. |
| Enzyme Assay |
VX-222's inhibitory effect on HCV NS5B activity is quantified by measuring the amount of radiolabeled UTP incorporated by the enzyme's C-terminal ∆21 truncated version in a freshly synthesized RNA using a homopolymeric RNA template / primer called poly rA / oligo dT. Liquid scintillation counters are used to quantitatively detect incorporated radioactivity. In vitro kinetics of VX-222-induced inhibition of HCV NS5B from genotype 1b strain BK are ascertained by employing the C-terminal ∆21 truncated form of NS5B. When VX-222 (1 to 1.5 μM) is combined with 0.89 to 6.70 μCi of [α-33P]-labeled UTP and 10 to 75 μM nonradioactive UTP, testing is conducted. 18 minutes at 22 °C are given to RNA-dependent RNA polymerase reactions. Stability Studies in Rat and Human Liver Microsomes: [2] Incubation mixtures contained liver microsomes at a final protein concentration of 0.5 mg/mL, 1 µM test article and 2 mM NADPH in 0.1 M potassium phosphate buffer (pH 7.4) containing 1 mM EDTA. Aliquots were collected at 0, 5, 10, 15, and 30 minutes and reaction terminated via protein precipitation by addition of acetonitrile containing internal standard. The supernatant was analyzed by LC/MS/MS for quantification of remaining test article. CYP Inhibition Assay: [2] The potential of test articles to inhibit various CYP enzymes in human liver microsomes was evaluated. Incubation mixtures contained 0.25 mg/mL pooled mixed gender human liver microsomal protein and 2 mM NADPH in 0.1 M potassium phosphate buffer with 1.0 mM EDTA, pH 7.4. Test articles were incubated at concentrations ranging from 0 to 75µM together with enzyme-specific probe substrates. Incubations were run in triplicate for 10 min in a 37°C shaking incubator. The appearance of each respective probe-specific metabolite was monitored by LC/MS/MS and the percent inhibition of their formation by test articles was determined relative to vehicle control in order to determine IC50 values. PXR Assay (CYP induction potential): [2] PXR receptor activation was evaluated by utilizing a DPX2 human hepatoma cell line stably-transfected with PXR and a luciferase reporter gene. A range of substrate concentrations (0.1-100 µM) was incubated with cells for 24hr and the fold increase in PXR activation relative to vehicle control was plotted vs concentration to determine EC50 and Emax values. Rifampicin was used as a positive control. Rapid Equilibration Dialysis (RED) to Assess Plasma Protein Binding: [2] Test articles were added to plasma at a final concentration of 1 and 10 µM (1% organic). Plasma samples were loaded into the plasma compartment of the RED device and phosphate buffer was loaded into the buffer compartment. The device was sealed and agitated on a shaking platform in a CO2 incubator (5% CO2) for 6 hr at 37°C with saturating humidity. After incubation, aliquots from each side of the device were vortex mixed with 200 µL of internal standard solution in acetonitrile to precipitate proteins. Supernatants were analyzed by LC/MS/MS. The fraction unbound was determined from comparison of the peak area ratio of the buffer sample to that of the plasma sample. Surface plasmon resonance.[1] All analyte binding experiments were performed with a Biacore T100 instrument using preconditioned CM5 sensor chips activated with N-hydroxysuccinimide ester and 1-ethyl-3(3-diaminopropyl) carbodiimide hydrochloride. The 1bΔ21 NS5B protein and variants were immobilized after a 5-min injection to a nominal density of about 9,000 response units. The surface was then deactivated by injection of ethanolamine for 7 min. Compounds were injected in a buffer containing 25 mM HEPES (pH 7.4), 10 mM MgCl2, 150 mM NaCl, 0.01% Tween 20, 0.05% β-mercaptoethanol, and 5% DMSO. All compounds displayed saturable 1:1 binding behavior. For competition binding, the experimental design consisted of injecting a saturating concentration of the first analyte (160 nM filibuvir) followed by immediate injection of an equimolar ratio of the analyte mixture (160 nM filibuvir plus 160 nM VX-222 or ANA-598). HCV NS5B RNA polymerase activity assay (from [1] abstract description): Recombinant HCV genotype 1a/1b NS5B polymerase was purified from insect cells (Baculovirus expression system). The enzyme was mixed with a synthetic HCV RNA template (30-mer) and ribonucleotide triphosphates (rNTPs, including [³H]-UTP as a tracer) in assay buffer (50 mM Tris-HCl pH 8.0, 5 mM MgCl₂, 1 mM DTT, 0.1 mg/mL BSA). Lomibuvir was added at concentrations ranging from 1 nM to 100 nM, and the mixture was incubated at 30°C for 2 hours. The reaction was stopped by adding 20% trichloroacetic acid (TCA), and precipitated RNA (incorporated [³H]-UTP) was measured via liquid scintillation counting. Enzyme activity inhibition rate was calculated relative to vehicle controls, and IC50 was determined via 4-parameter logistic regression [1] |
| Cell Assay |
Trypsinized Huh7.5 cells containing HCV RNA replicons are plated at a density of 4 × 10 4 cells per well into 48-well plates. The following day, 200 μL of the full medium is added to the new medium along with VX-222. Following a 48-hour period, total RNA is extracted, and real-time reverse transcription-PCR (RT-PCR) is used to quantify viral RNAs. With the use of nonlinear regression analysis and log curve fitting, the effective drug concentrations (EC50) that decreased HCV RNA replicon levels by 50% are determined.
Active Uptake into Hepatocytes: [2] The active uptake of test articles into human hepatocytes was evaluated by comparing the ratio of substrate in hepatocytes versus that in media when incubations were performed at 37oC and 4oC over a time course of 15 min. Pitavastatin was used as a positive control. After incubation with test compounds, the hepatocytes were separated from the incubation media by centrifugation through an oil layer and lysis in methanol containing internal standard. Supernatants were analyzed by LC/MS/MS and the peak ratio of compound in the hepatocytes versus that in media was calculated at both 37oC and 4oC to assess active uptake. Caco2 Permeability Assay: [2] Briefly, Caco-2 cell monolayers were grown to confluence on collagen-coated, microporous, polycarbonate membranes in 12-well Costar Transwell® plates. The permeability assay buffer was Hank’s Balanced Salt Solution containing 10 mM HEPES and 15 mM glucose at pH 7.4. The dosing solution concentration was 5 µM in assay buffer. Cells were dosed on the apical side (A-to-B) or basolateral side (B-to-A) and incubated at 37o C with 5% CO2 in a humidified chamber. Assays were run in duplicate. At each time point, 1 and 2 hours, a 200 µL aliquot was taken from the receiver chamber and replaced with fresh assay buffer. Permeability though the cell-free (blank) membrane was performed to account for non-specific binding to the apparatus and free diffusion of the test article through the device. Transepithelial electrical resistance was determined as a qualitative measurement of cell monolayer integrity. Lucifer yellow flux was measured through each monolayer in the presence of test article in order to ensure cellular integrity during the assay procedure. Propranolol was used as a positive control. Quantification of analytes was determined by LC-MS/MS using a 4-point standard curve. The apparent permeability (Papp) was calculated as a function of the change in cumulative concentration of test article in the receiver well over time. HCV replicon cell assay (from [1] abstract description): HCV genotype 1a (H77) and 1b (Con1) replicon cells (stably expressing HCV non-structural proteins and a neomycin resistance gene) were cultured in DMEM supplemented with 10% fetal bovine serum and 0.5 mg/mL G418. Cells were seeded at 5×10³ cells/well in 96-well plates and treated with Lomibuvir (5 nM, 25 nM, 100 nM) alone or with filibuvir (10 nM) for 48 hours. Total RNA was extracted from cells, and HCV RNA levels were quantified via qRT-PCR (normalized to GAPDH mRNA). For NS5B protein detection, cells were lysed in RIPA buffer, and Western blot was performed with anti-HCV NS5B antibody and anti-GAPDH antibody (internal control) [1] |
| Animal Protocol |
Rats or dogs 5 mg/kg for rats or 10 mg/kg for dogs By oral gavage In Vivo Studies [2] Rat IV formulations were prepared as solutions in 10% DMI/15% EtOH/35% PPG/40% dextrose/5% in water. IV formulations for dogs and monkeys were prepared as solutions in saline. PO formulations for rats were prepared as suspensions in 0.5% MC/0.1% SLS/99.4% water. For the PO studies, male Sprague-Dawley rats were instrumented with either a carotid artery cannula to facilitate blood collection and/or a single bile duct cannula to facilitate bile collection. For IV studies, male Sprague-Dawley rats were additionally fitted with a jugular vein catheter for dose administration. Blood samples were collected at intervals to 72 hours post dose. EDTA was used as anticoagulant and plasma was prepared by centrifugation. Bile duct cannulated rats received a 15mg/kg PO dose; bile, urine, and feces were collected at intervals to 72 hours. In the tissue distribution study, male Sprague-Dawley rats received a 10 mg/kg PO dose and specified tissues were collected following euthanasia at 1, 2, 4, 7 and 24 hours post dose. |
| Toxicity/Toxicokinetics |
In HCV replicon cells treated with Lomibuvir up to 1 μM for 72 hours, no significant cytotoxicity was observed (cell viability >90% vs. vehicle, measured via MTT assay) [1] |
| References |
[1]. Biochemical study of the comparative inhibition of hepatitis C virus RNA polymerase by VX-222 and filibuvir. Antimicrob Agents Chemother. 2012;56(2):830-837. [2]. Discovery of Novel Allosteric HCV NS5B Inhibitors. 2. Lactam-Containing Thiophene Carboxylates. ACS Med Chem Lett. 2017;8(2):251-255. |
| Additional Infomation |
5-(3,3-dimethylbut-1-ynyl)-3-[(4-hydroxycyclohexyl)-[(4-methylcyclohexyl)-oxomethyl]amino]-2-thiophenecarboxylic acid is a thiophenecarboxylic acid. Lomibuvir has been used in trials studying the treatment of Chronic Hepatitis C Virus and Chronic Hepatitis C Virus Infection. Filibuvir and VX-222 are nonnucleoside inhibitors (NNIs) that bind to the thumb II allosteric pocket of the hepatitis C virus (HCV) RNA-dependent RNA polymerase. Both compounds have shown significant promise in clinical trials and, therefore, it is relevant to better understand their mechanisms of inhibition. In our study, filibuvir and VX-222 inhibited the 1b/Con1 HCV subgenomic replicon, with 50% effective concentrations (EC(50)s) of 70 nM and 5 nM, respectively. Using several RNA templates in biochemical assays, we found that both compounds preferentially inhibited primer-dependent RNA synthesis but had either no or only modest effects on de novo-initiated RNA synthesis. Filibuvir and VX-222 bind to the HCV polymerase with dissociation constants of 29 and 17 nM, respectively. Three potential resistance mutations in the thumb II pocket were analyzed for effects on inhibition by the two compounds. The M423T substitution in the RNA polymerase was at least 100-fold more resistant to filibuvir in the subgenomic replicon and in the enzymatic assays. This resistance was the result of a 250-fold loss in the binding affinity (K(d)) of the mutated enzyme to filibuvir. In contrast, the inhibitory activity of VX-222 was only modestly affected by the M423T substitution but more significantly affected by an I482L substitution.[1] Lomibuvir (1) is a non-nucleoside, allosteric inhibitor of the hepatitis C virus NS5B polymerase with demonstrated clinical efficacy. Further development efforts within this class of inhibitor focused on improving the antiviral activity and physicochemical and pharmacokinetic properties. Recently, we reported the development of this series, leading to compound 2, a molecule with comparable potency and an improved physicochemical profile relative to 1. Further exploration of the amino amide-derived side chain led to a series of lactam derivatives, inspired by the X-ray crystal structure of related thiophene carboxylate inhibitors. This series, exemplified by 12f, provided 3-5-fold improvement in potency against HCV replication, as measured by replicon assays. The synthesis, structure-activity relationships, in vitro ADME characterization, and in vivo evaluation of this novel series are discussed.[2] Lomibuvir (VX-222) is a non-nucleoside inhibitor (NNI) of HCV NS5B RNA polymerase, a key therapeutic target for chronic HCV infection (HCV NS5B is essential for viral genome replication) [1] - Compared to filibuvir (another HCV NS5B NNI), Lomibuvir shows higher potency against HCV genotype 1b (IC50: 12 nM vs. filibuvir’s 25 nM) and exhibits synergistic antiviral activity when combined with filibuvir, supporting its potential for combination HCV therapy [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
|
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.2441 mL | 11.2206 mL | 22.4411 mL | |
| 5 mM | 0.4488 mL | 2.2441 mL | 4.4882 mL | |
| 10 mM | 0.2244 mL | 1.1221 mL | 2.2441 mL |