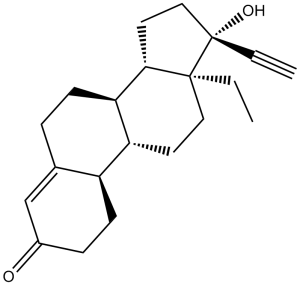
Levonorgestrel (Norgestrel, Microval, Postinor, Mirena, Plan B) is a female hormone that can prevent ovulation and has been used in many birth control pills. Levonorgestrel is a synthetic progestin that binds to progesterone and androgen receptors/AR but not the estrogen receptor/ER. It induces apoptosis in ovarian epithelium cells. Levonorgestrel suppresses the stimulation of progesterone secretion induced by oLH, dibutyryl-cAMP and Pregnenolone in rats luteal cells. Levonorgestrel also inhibits constrictions evoked by either a high potassium (K(+)) solution or phorbol myristate acetate (PMA) in the absence and presence of extracellular calcium (Ca(2+)). Levonorgestrel is useful within 120 hours as emergency birth control. It becomes less effective the longer after sex and only works before pregnancy has occurred. It is also combined with an estrogen to make combined oral birth control pill.
Physicochemical Properties
| Molecular Formula | C21H28O2 | |
| Molecular Weight | 312.45 | |
| Exact Mass | 312.208 | |
| CAS # | 797-63-7 | |
| Related CAS # | Dydrogesterone;152-62-5;Levonorgestrel-d8;Norgestrel-d6;2376035-98-0 | |
| PubChem CID | 13109 | |
| Appearance | White to off-white solid powder | |
| Density | 1.1±0.1 g/cm3 | |
| Boiling Point | 459.1±45.0 °C at 760 mmHg | |
| Melting Point | 206ºC | |
| Flash Point | 195.4±21.3 °C | |
| Vapour Pressure | 0.0±2.6 mmHg at 25°C | |
| Index of Refraction | 1.571 | |
| LogP | 3.92 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 2 | |
| Rotatable Bond Count | 2 | |
| Heavy Atom Count | 23 | |
| Complexity | 609 | |
| Defined Atom Stereocenter Count | 6 | |
| SMILES | CC[C@]12CC[C@H]3[C@H]([C@@H]1CC[C@]2(C#C)O)CCC4=CC(=O)CC[C@H]34 |
|
| InChi Key | WWYNJERNGUHSAO-XUDSTZEESA-N | |
| InChi Code | InChI=1S/C21H28O2/c1-3-20-11-9-17-16-8-6-15(22)13-14(16)5-7-18(17)19(20)10-12-21(20,23)4-2/h2,13,16-19,23H,3,5-12H2,1H3/t16-,17+,18+,19-,20-,21-/m0/s1 | |
| Chemical Name | (8R,9S,10R,13S,14S,17R)-13-ethyl-17-ethynyl-17-hydroxy-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3(2H)-one | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| ln Vitro | Levonorgestrel (5-25 mg/mL; 72 h) has the concentration-dependent ability to suppress cell growth and increase apoptosis in uterine leiomyoma cells [1]. Levonorgestrel (0.1-100 μM; 4 h) can decrease the generation of progesterone at high concentrations (100 μM) in luteal cells, while it has no effect at low doses (0-10 μM) [2]. |
| ln Vivo | In Sprague–Dawley rats, levonorgestrel (0.005-0.15 mg/kg; once every two days for three weeks) decreases bone turnover, inhibits bone resorption, and raises bone mineral content [3]. Apodemus agrarius mice can successfully avoid pregnancy when levonorgestrel (1 mg/kg; gavage; once daily for three consecutive days) is combined with ethinyl estradiol [4]. |
| Cell Assay |
Western Blot Analysis[1] Cell Types: Uterine leiomyoma cells Tested Concentrations: 5 mg/mL; 10mg/mL; 20 mg/mL Incubation Duration: Experimental Results: Inhibited Bcl-2 and survivin expression at high concentrations (10 mg/mL and 20 mg /mL). Dramatically increased the phosphorylation of P38 phosphorylation and increased Caspase-3 expression at high concentrations (10 mg/mL and 20 mg/mL). |
| Animal Protocol |
Animal/Disease Models: Apodemus agrarius model[4] Doses: 1 mg/kg Route of Administration: intragastric (po) administration (ig), one time/day for three days Experimental Results: Damaged the sperm ducts, decreased sperm production in combination with quinestrol. decreased population density in the field in combination with quinestrol. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Norgestrel is absorbed from the gastrointestinal tract, metabolised by the liver and excreted in the urine and faeces as glucuronide and sulphate conjugates. (14)C-Norgestrel was administered to seven human subjects and 43% of dose was excreted in the urine within 5 days; the biological half-life of the radioactivity was 24 hr. Enzymic hydrolysis released only 32% of the urinary radioactivity and a further 25% was excreted as sulphate conjugates. The metabolites excreted in the urine were much less polar than those following the administration of the related compounds, norethisterone or lynestrenol. The 3alphaOH,5beta and 3betaOH,5beta isomers of the tetrahydronorgestrel (13beta-ethyl-17alpha-ethynyl-5 beta-gonane-3alpha,17beta-diol) were isolated from urine and identified by mass spectrometry and thin-layer and gas-liquid chromatography. Plasma radioactivity decreased more rapidly than after the administration of norethisterone and lynestrenol. About 2% of the administered dose was converted to acidic compounds. There was no apparent difference in the rate of excretion of radioactivity or in the metabolites after either oral or intravenous administration of norgestrel. The binding of different synthetic steroids, used in hormonal contraception, to Sex Hormone Binding Globulin (SHBG) was studied by measuring their ability to displace tritiated testosterone from SHBG in a competitive protein binding system. Only 19-nortestosterone derivates had any significant ability to displace testosterone from SHBG, d-norgestrel (d-Ng) being the strongest displacer. Increasing the SHBG levels in women with previous constant plasma d-Ng levels increased these levels two- to sixfold. It is concluded that SHBG is the main carrier protein for d-Ng. The strong testosterone displacing activity of d-Ng might also explain androgenic side effects observed with d-Ng containig oral contraceptives. Metabolism / Metabolites (14)C-Norgestrel was administered to seven human subjects and 43% of dose was excreted in the urine within 5 days ... Enzymic hydrolysis released only 32% of the urinary radioactivity and a further 25% was excreted as sulphate conjugates. The metabolites excreted in the urine were much less polar than those following the administration of the related compounds, norethisterone or lynestrenol. The 3alphaOH,5beta and 3betaOH,5beta isomers of the tetrahydronorgestrel (13beta-ethyl-17alpha-ethynyl-5 beta-gonane-3alpha,17beta-diol) were isolated from urine and identified by mass spectrometry and thin-layer and gas-liquid chromatography. Plasma radioactivity decreased more rapidly than after the administration of norethisterone and lynestrenol. About 2% of the administered dose was converted to acidic compounds. There was no apparent difference in the rate of excretion of radioactivity or in the metabolites after either oral or intravenous administration of norgestrel. The comparative metabolism of dl-, d-, and l-norgestrel was investigated in African Green Monkeys (Cercopithecus aethiops). Total (14)C excretion in urine after a single oral dose of (14)C-dl-norgestrel (1 mg/kg) was significantly higher (51.4 +/- 5.0%) than that observed after administration of the d-enantiomer (37.5 +/- 5.4%) but not the l-enantiomer (44.2 +/- 8.9%). In all cases, the major part of the urinary radioactivity was present in a free fraction (48-62%), while an additional 13-27% was released by beta-glucuronidase preparations. No sulfate conjugates were detected. At least one major (16beta-hydroxylation) and one minor (16alpha-hydroxylation) metabolic pathway were stereoselective, i.e., they are operative with the I-but not the d-enantiomer. Three metabolites, 16beta-hydroxynorgestrel, 16alpha-hydroxynorgestrel, and 16-hydroxytetrahydronorgestrel (believed to be 16beta) were only detected in urine samples obtained from (14)C-dland -l-norgestrel-dosed animals. Following (14)C-d-norgestrel administration, 3alpha, 5beta-tetrahydronorgestrel was found to be the major urinary metabolite. These observations are compared with those reported earlier on the urinary metabolites of dl-norgestrel in women. The in vitro metabolism of stereo-isomers (d, l and the racemic mixture dl) of norgestrel by a microsomal fraction from rabbit liver was investigated. The metabolism of the biologically active l-norgestrel was more rapid than that of d-norgestrel (sic.) which is biologically inactive. This was mainly due to the more ready conversion of l-norgestrel to ring-A reduced metabolites. There was no difference between the two isomers in respect of the amount undergoing hydroxylation; about 40% of each isomer was converted to hydroxylated metabolites after 30 min incubation. However, there were differences between the isomers, l-norgestrel being converted mainly to the 16beta-hydroxysteroid and d-norgestrel to the 16alpha-hydroxysteroid. Similar amounts of both isomers were hydroxylated at C-6. The metabolism of the racemic mixture was intermediate between that of the d and l isomers. The rates of metabolism of synthetic gestagens derived from 19-nortestosterone by rabbit liver tissue in vitro were compared. Over a period of 1 hr norethisterone was metabolized as rapidly as 19-nortestosterone whereas d-norgestrel and lynestrenol were metabolized at a slightly lower rate. Less than 5% of l-norgestrel was metabolized. In all cases the reaction product was the tetrahydrosteroid. Lynestrenol was metabolised through norethisterone. Skeletal muscle, lung and small intestine also metabolized norethisterone and d-norgestrel but at a slower rate than liver tissue. Small amounts of norethisterone were metabolized by adipose tissue but heart and spleen were inactive. Lynestrenol and l-norgestrel were not metabolized by any of the extra-hepatic tissues studied. In vitro studies were conducted on the metabolism of 3 steroids used in OCs (oral contraceptives) by small pieces of human jejunal mucosa. This was done because the gastrointestinal mucosa of humans is known to metabolize a number of drugs. Almost 40% of the ethinyl estradiol, 9.8% of the levonorgestrel, and 7% of the mestranol were metabolized after incubation. All these metabolic responses were significantly different from those in the control groups. Results of the study show that the metabolism of the ethinyl estradiol was related to the weight of the tissue used. These results are consistent with the known marked 1st pass effect of ethinyl estradiol. Norgestrel, known to have little or no 1st pass effect, did not show a high rate of gut metabolism. Under the experimental conditions employed, no Phase 1 metabolism of either ethinyl estradiol or levonorgestrel was apparent. Hepatic. Route of Elimination: About 45% of levonorgestrel and its metabolites are excreted in the urine and about 32% are excreted in feces, mostly as glucuronide conjugates. Biological Half-Life (14)C-Norgestrel was administered to seven human subjects and 43% of dose was excreted in the urine within 5 days; the biological half-life of the radioactivity was 24 hr. ... |
| Toxicity/Toxicokinetics |
Toxicity Summary Binds to the progesterone and estrogen receptors. Target cells include the female reproductive tract, the mammary gland, the hypothalamus, and the pituitary. Once bound to the receptor, progestins like levonorgestrel will slow the frequency of release of gonadotropin releasing hormone (GnRH) from the hypothalamus and blunt the pre-ovulatory LH (luteinizing hormone) surge. Toxicity Data LD50 >5000 mg/kg (orally in rats) Interactions The metabolism of estrogens and progestagens may be increased by concomitant use of substances known to induce drug-metabolising enzymes, specifically cytochrome P450 enzymes, such as anticonvulsants (eg phenobarbital, phenytoin, carbamazepine), and anti-infectives (eg rifampicin, rifabutin, nevirapine, efavirenz). Ritonavir and nelfinavir, although known as strong inhibitors, by contrast exhibit inducing properties when used concomitantly with steroid hormones. Herbal preparations containing St John's Wort (Hypericum Perforatum) may induce the metabolism of estrogens and progestagens. Phenytoin and rifampin increase the serum concentrations of sex hormone-binding globulin (SHBG); this significantly decreases the serum concentration of free drug for some progestins, which is a special concern in patients using progestins for contraception. /Progestins/ Drug interaction data are not available for rifabutin, but because its structure is similar to that of rifampin, similar precautions with its use with progestins may be warranted. ... /Progestins/ Non-Human Toxicity Values LD50 Rat oral 5010 mg/kg LD50 Rat ip 11,200 mg/kg LD50 Mouse ip 7300 mg/kg LD50 Mouse oral 5010 mg/kg |
| References |
[1]. Levonorgestrel inhibits proliferation and induces apoptosis in uterine leiomyoma cells. Contraception vol. 82,3 (2010): 301-8. [2]. Levonorgestrel inhibits luteinizing hormone-stimulated progesterone production in rat luteal cells. The Journal of steroid biochemistry and molecular biology vol. 50,3-4 (1994): 161-6. [3]. Effects of different nylestriol/levonorgestrel dosages on bone metabolism in female Sprague-Dawley rats with retinoic acid-induced osteoporosis. Endocrine research vol. 29,1 (2003): 23-42. [4]. Anti-fertility effect of levonorgestrel and/or quinestrol on striped field mouse (Apodemus agrarius): evidence from both laboratory and field experiments. Integrative zoology vol. 17,6 (2022): 1041-1052. [5]. Effects of oral and vaginal administration of levonorgestrel emergency contraception on markers of endometrial receptivity. Human reproduction (Oxford, England) vol. 25,4 (2010): 874-83. |
| Additional Infomation |
Therapeutic Uses Contraceptives, Oral, Synthetic; Progestational Hormones, Synthetic Low-ogestrel (norgestrel and ethinyl estradiol tablets) is indicated for the prevention of pregnancy in women who elect to use this product as a method of contraception. /Included in US product label/ /Cyclo-Progynova is indicated as/ hormone replacement therapy (HRT) for estrogen deficiency symptoms in perimenopausal and postmenopausal women. /Cyclo-Progynova is indicated for/ prevention of osteoporosis in postmenopausal women at high risk of future fractures who are intolerant of, or contraindicated for, other medicinal products approved for the prevention of osteoporosis. Norgestrel ... /is/ indicated for the prevention of pregnancy. Progestin-only oral contraceptives are also called minipills and progestin-only oral pills (POPs). /Former/ Drug Warnings Cigarette smoking increases the risk of serious cardiovascular side effects from oral contraceptive use. This risk increases with age and with heavy smoking (15 or more cigarettes per day) and is quite marked in women over 35 years of age. Women who use oral contraceptives should be strongly advised not to smoke. The use of oral contraceptives is associated with increased risks of several serious conditions including myocardial infarction, thromboembolism, stroke, hepatic neoplasia, and gallbladder disease, although the risk of serious morbidity or mortality is very small in healthy women without underlying risk factors. The risk of morbidity and mortality increases significantly in the presence of other underlying risk factors such as hypertension, hyperlipidemias, hypercholesterolemia, obesity and diabetes. Oral contraceptives should not be used in women who have the following conditions: thrombophlebitis or thromboembolic disorders; a past history of deep vein thrombophlebitis or thromboembolic disorders; cerebral vascular or coronary artery disease; Known or suspected carcinoma of the breast; carcinoma of the endometrium or other known or suspected estrogen-dependent neoplasia; undiagnosed abnormal genital bleeding; cholestatic jaundice of pregnancy or jaundice with prior pill use; hepatic adenomas, carcinomas or benign liver tumors; known or suspected pregnancy The most frequent adverse effect of oral contraceptives is nausea. In addition, nausea has been reported in women using vaginal or transdermal estrogen-progestin contraceptives. The principal risk associated with currently recommended high-dose, postcoital estrogen-progestin combination regimens appears to be moderate to severe adverse GI effects including severe vomiting and nausea, which occur in 12-22 and 30-66%, respectively, of women receiving the short-course regimens and may limit compliance with, and effectiveness of, the regimens. In 2 prospective, randomized studies, nausea and vomiting were less common with a high-dose postcoital progestin-only regimen (0.75 mg levonorgestrel every 12 hours for 2 doses) than with a high-dose estrogen-progestin regimen (100 mcg ethinyl estradiol and 0.5 mg levonorgestrel every 12 hours for 2 doses). Other adverse GI effects include vomiting, abdominal cramps, abdominal pain, bloating, diarrhea, and constipation. Gingivitis and dry socket have also been reported. Changes in appetite and changes in weight also may occur. /Estrogen-Progestin Combination/ For more Drug Warnings (Complete) data for NORGESTREL (52 total), please visit the HSDB record page. |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (8.00 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2005 mL | 16.0026 mL | 32.0051 mL | |
| 5 mM | 0.6401 mL | 3.2005 mL | 6.4010 mL | |
| 10 mM | 0.3201 mL | 1.6003 mL | 3.2005 mL |