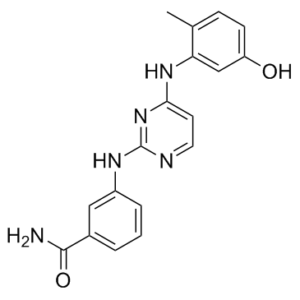
Lck inhibitor 2, a bis-anilinopyrimidine derivative, is a novel and potent multi-kinase inhibitor of tyrosine kinases including LCK, BTK, LYN, SYK, and TXK with IC50 values of 13nM, 9nM, 3nM, 26nM and 2nM for Lck, Btk, Lyn, Btk and Txk respectively.
Physicochemical Properties
| Molecular Formula | C18H17N5O2 |
| Molecular Weight | 335.35988 |
| Exact Mass | 335.138 |
| Elemental Analysis | C, 64.47; H, 5.11; N, 20.88; O, 9.54 |
| CAS # | 944795-06-6 |
| Related CAS # | Lck Inhibitor;847950-09-8 |
| PubChem CID | 25138012 |
| Appearance | Light yellow to yellow solid powder |
| LogP | 3.455 |
| Hydrogen Bond Donor Count | 4 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 25 |
| Complexity | 449 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | O=C(C1C=C(NC2N=C(NC3C(C)=CC=C(O)C=3)C=CN=2)C=CC=1)N |
| InChi Key | SFCBIFOAGRZJNX-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C18H17N5O2/c1-11-5-6-14(24)10-15(11)22-16-7-8-20-18(23-16)21-13-4-2-3-12(9-13)17(19)25/h2-10,24H,1H3,(H2,19,25)(H2,20,21,22,23) |
| Chemical Name | 3-[[4-(5-hydroxy-2-methylanilino)pyrimidin-2-yl]amino]benzamide |
| Synonyms | Lck Inhibitor 2; Lck Inhibitor-2 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Lymphocyte-specific kinase (Lck) (IC50 = 13 nM, Table 1)[2] Bruton's tyrosine kinase (Btk) (IC50 = 39 nM, Table 4)[2] Lyn (IC50 = 26 nM, Table 4)[2] Syk (IC50 = 23 nM, Table 4)[2] Tsk (IC50 = 7 nM, Table 4)[2] |
| ln Vitro |
Inhibition of leukocyte-specific protein tyrosine kinase (Lck) activity offers one of the approaches for the treatment of T-cell mediated inflammatory disorders including rheumatoid arthritis, transplant rejection and inflammatory bowel disease.To explore the relationship between the structures of the N-4 Pyrimidinyl-1H-indazol-4-amines and their Lck inhibition, 3D-QSAR study using CoMFA analysis have been performed on a dataset of 42 molecules. The bioactive conformation of the template molecule, selected as the most potent molecule 23 from the series was obtained by performing molecular docking at the ATP binding site of Lck, which is then used to build the rest of the molecules in the series. [3] In an enzyme inhibition assay, compound 2 potently inhibited Lck kinase activity with an IC50 of 13 nM (Table 1). It also showed appreciable activity against other tyrosine kinases: Btk (IC50 39 nM), Lyn (IC50 26 nM), Syk (IC50 23 nM), and Tsk (IC50 7 nM) (Table 4).[2] Molecular docking and modeling studies predicted that 2 binds to the ATP-binding site of Lck. The pyrimidine core forms two typical hydrogen bonds with the hinge region (backbone NH and carbonyl of Met319). The 2-methyl substituent on the 4-aniline ring occupies a small lipophilic pocket formed by Val259, Ala271, and Lys273. The 5-hydroxyl group is predicted to form a hydrogen bond with the backbone NH of Asp382 and potentially a direct or water-mediated interaction with the sidechain of Glu288 (Fig. 1).[2] |
| Enzyme Assay |
Lck kinase inhibition activity was assessed using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay. Murine Lck was overexpressed in insect cells and used as a semi-purified lysate. The enzyme was pre-activated at room temperature for 30 minutes in a buffer containing 10 mM MgCl2 and 100 µM ATP. The activated Lck was then diluted 100-fold in 100 mM HEPES buffer, pH 7.4. For the assay, 15 µL of the diluted, activated Lck was added to wells of a 384-well plate containing 1 µL of serially diluted test compound or DMSO vehicle (final DMSO concentration 3.3%). The mixture was incubated for 15 minutes at room temperature. The kinase reaction was initiated by adding 15 µL of a substrate reagent mixture containing biotinylated peptide substrate (Biotin-EEEEYFELV, final concentration 0.5 µM), ATP (final concentration 120 µM), and MgCl2 (final concentration 20 mM) in 100 mM HEPES buffer, pH 7.4. The reaction mixture was incubated at room temperature for 60 minutes. The reaction was stopped by adding 30 µL of a detection reagent containing EDTA, a europium-labeled anti-phosphotyrosine antibody, and streptavidin-allophycocyanin. After a further incubation period, the TR-FRET signal was measured, and IC50 values were calculated from dose-response curves.[2] |
| ADME/Pharmacokinetics |
The pharmacokinetic parameters of the phenol analogue 23 (a close structural analogue of compound 2 with a sulfonamide at the 2-position but the same 4-(2-methyl-5-hydroxyaniline) substituent) were determined in female Balb/C mice and served as a reference for the phenol series, which includes 2. After intravenous (IV) administration of 23 at 2.5 mg/kg, it exhibited high plasma clearance (CLp = 65.5 mL/min/kg), a low volume of distribution (Vd = 0.3 L/kg), and a very short half-life (T1/2 = 0.12 hours). After oral (PO) administration at 10 mg/kg, the oral bioavailability (F%) was less than 1%. This poor pharmacokinetic profile was attributed to the metabolic lability of the phenol moiety.[2] |
| References |
[1]. Assessment of Chemical Coverage of Kinome Space and Its Implications for Kinase Drug Discovery. Journal of Medicinal Chemistry (2008), 51(24), 7898-7914. [2]. N-4-Pyrimidinyl-1H-indazol-4-amine inhibitors of Lck: Indazoles as phenol isosteres with improved pharmacokinetics. Bioorganic & Medicinal Chemistry Letters (2007), 17(15), 4363-4368. [3]. Molecular docking guided 3D-QSAR CoMFA analysis of N-4-Pyrimidinyl-1H-indazol-4-amine inhibitors of leukocyte-specific protein tyrosine kinase. Journal of Molecular Modeling (2008), 14(10), 937-947. |
| Additional Infomation |
Compound 2 is a 2,4-dianilinopyrimidine derivative identified as a potent inhibitor of Lck, a key tyrosine kinase in T-cell receptor signaling. It was designed based on the scaffold of known kinase inhibitors, incorporating a 4-(2-methyl-5-hydroxyaniline) substituent which was critical for high potency.[2] Despite its high in vitro potency against Lck, compounds containing the 5-hydroxyaniline (phenol) moiety, like 2, exhibited poor pharmacokinetic properties in mice (high clearance, short half-life, low oral bioavailability), likely due to rapid phase II metabolism of the phenol group. This limitation prompted the search for phenol isosteres, leading to the discovery of indazole-based replacements (e.g., compound 37) with retained potency and significantly improved PK profiles.[2] The binding mode of 2, as proposed by molecular modeling and supported by later structural studies on related compounds, involves key interactions: hinge binding via the pyrimidine, lipophilic filling of a pocket by the 2-methyl group, and a dual hydrogen-bonding role for the 5-hydroxyl with Asp382 and Glu288 in the active site of Lck.[2] |
Solubility Data
| Solubility (In Vitro) | DMSO: ~20 mg/mL (~59.6 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2 mg/mL (5.96 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2 mg/mL (5.96 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2 mg/mL (5.96 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.9819 mL | 14.9094 mL | 29.8187 mL | |
| 5 mM | 0.5964 mL | 2.9819 mL | 5.9637 mL | |
| 10 mM | 0.2982 mL | 1.4909 mL | 2.9819 mL |