LXR623 (WAY-252623; LXR-623) is a novel, highly brain-penetrant, selective and orally bioavailable synthetic modulator of LXR (Liver X receptor) with anticancer activity, It is a partial LXRα and full LXRβ agonist with IC50s of 24 nM and 179 nM, respectively. As a LXRα-partial/LXRβ-full agonist, LXR623 selectively kills GBM cells in an LXRβ- and cholesterol-dependent fashion, causing tumor regression and prolonged survival in mouse models. LXR623 displayed high efficacy in reducing lesion progression in the murine LDLR(-/-) atherosclerosis model with no associated increase in hepatic lipogenesis either in this model or Syrian hamsters. LXR-623 potently kills U87EGFRvIII and GBM39 cells in vitro while completely sparing NHAs. LXR-623 also increases ABCA1 protein and decreases LDLR protein levels in all three cell lines.
Physicochemical Properties
| Molecular Formula | C21H12CLF5N2 | |
| Molecular Weight | 422.78 | |
| Exact Mass | 422.06 | |
| CAS # | 875787-07-8 | |
| Related CAS # |
|
|
| PubChem CID | 16734800 | |
| Appearance | White to off-white solid powder | |
| Density | 1.4±0.1 g/cm3 | |
| Boiling Point | 528.4±50.0 °C at 760 mmHg | |
| Melting Point | 100 °C | |
| Flash Point | 273.4±30.1 °C | |
| Vapour Pressure | 0.0±1.3 mmHg at 25°C | |
| Index of Refraction | 1.583 | |
| LogP | 6.05 | |
| Hydrogen Bond Donor Count | 0 | |
| Hydrogen Bond Acceptor Count | 6 | |
| Rotatable Bond Count | 3 | |
| Heavy Atom Count | 29 | |
| Complexity | 554 | |
| Defined Atom Stereocenter Count | 0 | |
| InChi Key | KYWWJENKIMRJBI-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C21H12ClF5N2/c22-18-10-15(24)9-6-13(18)11-29-20(12-4-7-14(23)8-5-12)16-2-1-3-17(19(16)28-29)21(25,26)27/h1-10H,11H2 | |
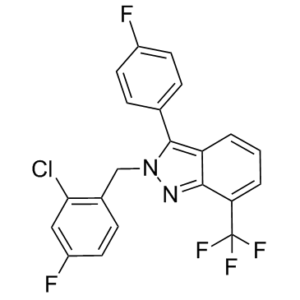
| Chemical Name | 2-[(2-chloro-4-fluorophenyl)methyl]-3-(4-fluorophenyl)-7-(trifluoromethyl)indazole | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
LXR623 targets human liver X receptor α (LXRα) (EC50 = 15 nM for transcriptional activation in reporter gene assay) [3] LXR623 targets human liver X receptor β (LXRβ) (EC50 = 22 nM for transcriptional activation in reporter gene assay) [3] LXR623 exhibits high selectivity for LXRα/β, no significant binding to other nuclear receptors (e.g., PPARγ, RXRα, FXR) at concentrations up to 1 μM [3][4] |
| ln Vitro |
In vitro, LXR-623 completely protects NHA while killing U87EGFRvIII and GBM39 cells efficiently. In all three cell lines, LXR-623 also caused an increase in ABCA1 protein and a decrease in LDLR protein levels. In every GBM sample examined, LXR-623 caused massive cell death, upregulated the expression of the ABCA1 efflux transporter, and inhibited the expression of LDLR. By activating LXRβ, LXR-623 (5 μM) also causes GBM cell death[1]. When human PBMCs were treated with LXR-623 in vitro, the transcription of ABCA1 and ABCG1 was markedly elevated [4]. In LXR reporter gene assay (HEK293 cells transfected with LXRα/β and LXR-responsive luciferase plasmid), LXR623 dose-dependently activated LXRα (EC50 = 15 nM) and LXRβ (EC50 = 22 nM), with maximal activation (~3.8-fold for LXRα, ~3.2-fold for LXRβ) at 100 nM [3] - In U87MG and U251 (glioblastoma, GBM) cells, LXR623 (1-10 μM) dose-dependently inhibited cell proliferation: IC50 = 4.2 μM (U87MG) and 5.8 μM (U251) (72-hour MTT assay). 10 μM treatment reduced colony-forming efficiency by ~65% (U87MG) and ~58% (U251) [1] - Quantitative PCR (qPCR) in GBM cells showed LXR623 (5 μM, 24 hours) upregulated LXR target genes involved in cholesterol efflux: ABCA1 mRNA by ~4.5-fold, ABCG1 by ~3.8-fold, and APOE by ~3.2-fold (U87MG cells); similar trends were observed in U251 cells [1] - In human peripheral blood mononuclear cells (PBMCs), LXR623 (0.1-5 μM) dose-dependently induced transcriptional biomarkers of LXR activation: 5 μM increased ABCA1 mRNA by ~5.2-fold, ABCG1 by ~4.8-fold, and SREBP-1c by ~3.5-fold (qPCR) [4] - LXR623 (up to 20 μM) did not affect the viability of normal human astrocytes or PBMCs (CC50 > 20 μM) [1][4] - In cholesterol-depleted GBM cells, LXR623 (5 μM) further enhanced cholesterol efflux to apolipoprotein A-I (apoA-I) by ~70%, reducing intracellular cholesterol levels by ~45% (radiolabeled cholesterol efflux assay) [1] |
| ln Vivo |
With little peripheral activity, LXR-623 (400 mg/kg, po) penetrates the blood-brain barrier, triggers the expression of the target gene, and reaches processing levels in GBM cells in the brain. LXR-623 prolongs the survival of mice with intracranial patient-derived GBMs by suppressing tumor growth and promoting tumor cell death [1]. When compared to a placebo, LXR-623 (1.5, 5 mg/kg/day) dramatically slowed the advancement of atherosclerosis in animals [2]. A dose-dependent manner was observed in the significant reduction of atherosclerosis by WAY-252623 (15 and 50 mg/kg). In Syrian hamsters that express CETP, WAY-252623 (20, 60, and 120 mg/kg/day, po) exhibits neutral lipid effects [3]. Additionally, rats' peripheral blood cells showed increased gene expression in response to LXR-623 (50 mg/kg). In monkey whole blood cells, LXR-623 (0, 15 and 50 mg/kg) dose-dependently and proportionately increases the transcription of ABCA1 and ABCG1 [4]. Antitumor efficacy in GBM xenograft model: Female BALB/c nude mice bearing U87MG glioblastoma xenografts were treated with LXR623 (50 mg/kg/day, 100 mg/kg/day, oral) for 28 days. High-dose treatment achieved a tumor growth inhibition (TGI) rate of 68%, reducing tumor weight from 1.52 ± 0.18 g (vehicle) to 0.49 ± 0.07 g. Tumor tissue analysis showed increased ABCA1 and APOE protein levels (~2.8-fold and ~2.3-fold, respectively) and reduced intracellular cholesterol content (~40%) [1] - Atherosclerosis regression in apoE-/- mice: Male apoE-/- mice fed a high-fat diet were treated with LXR623 (30 mg/kg/day, oral) for 12 weeks. Magnetic resonance imaging (MRI) showed a ~55% reduction in aortic plaque volume compared to vehicle. Plaque analysis revealed increased collagen content (~60%) and reduced necrotic core area (~45%), improving plaque stability [2] - Lipid-lowering effect in primates: Male rhesus monkeys with diet-induced hyperlipidemia were treated with LXR623 (10 mg/kg/day, oral) for 4 weeks. Serum LDL cholesterol was reduced by ~32%, total cholesterol by ~25%, and triglycerides by ~18% compared to baseline. No significant increase in hepatic triglyceride content was observed [3] - Lipid-neutral effect in hamsters: Male Golden Syrian hamsters were treated with LXR623 (1-10 mg/kg/day, oral) for 2 weeks. Unlike other LXR agonists, LXR623 did not increase serum triglycerides or hepatic lipid accumulation at doses up to 10 mg/kg, maintaining lipid neutrality [3] - No significant weight loss or overt toxicity (lethargy, organ damage, hematological/biochemical abnormalities) was observed in treated animals [1][2][3] |
| Enzyme Assay |
LXR reporter gene assay: HEK293 cells were co-transfected with human LXRα or LXRβ expression plasmid, LXR-responsive luciferase reporter plasmid (containing LXR response elements, LXREs), and β-actin-renilla plasmid (internal control). After 24-hour transfection, cells were treated with serial dilutions of LXR623 (0.01-1000 nM) for 24 hours. Luciferase activity was measured using a dual-luciferase assay system, and relative luciferase activity (firefly/renilla) was calculated to assess LXR activation. EC50 values were derived from dose-response curves [3][4] - Cholesterol efflux assay: U87MG cells were labeled with [³H]cholesterol for 24 hours, then incubated with LXR623 (0.1-10 μM) in the presence of apoA-I (cholesterol acceptor) for 12 hours. Radioactivity in the medium (effluxed cholesterol) and cell lysates (remaining cholesterol) was measured by liquid scintillation counting. Cholesterol efflux rate was calculated as (medium radioactivity / total radioactivity) × 100% [1] |
| Cell Assay |
GBM cell antiproliferation assay: U87MG and U251 cells were seeded in 96-well plates (5×10³ cells/well) and allowed to attach for 24 hours. Serial dilutions of LXR623 (0.1-20 μM) were added, and cells were cultured for 72 hours. MTT reagent was added, and absorbance at 570 nm was measured to calculate cell viability and IC50 values [1] - Colony-forming assay: U87MG and U251 cells were seeded in 6-well plates (200 cells/well) and treated with LXR623 (1-10 μM) for 14 days. Colonies were fixed with methanol, stained with crystal violet, and counted. Colony-forming efficiency was calculated as the percentage of colonies formed relative to vehicle control [1] - LXR target gene expression assay: GBM cells or PBMCs were seeded in 6-well plates (2×10⁵ cells/well) and treated with LXR623 (0.1-5 μM) for 24 hours. Total RNA was extracted, reverse-transcribed to cDNA, and qPCR was performed to quantify mRNA levels of ABCA1, ABCG1, APOE, and SREBP-1c. GAPDH was used as a reference gene for normalization [1][4] - Normal cell viability assay: Normal human astrocytes and PBMCs were seeded in 96-well plates (5×10³ cells/well) and treated with LXR623 (0.1-20 μM) for 72 hours. MTT reagent was added, and absorbance at 570 nm was measured to calculate cell viability [1][4] |
| Animal Protocol |
Dissolved in 0.5% methylcellulose, 2% Tween 80 in water; 400 mg/kg; Oral gavage Five-week-old female athymic nu/nu mice with U87EGFRvIII IRFP720 or GBM39 IRFP720 cells intracranially injected into the mouse brain. U87MG GBM xenograft model: Female BALB/c nude mice (4-6 weeks old) were subcutaneously implanted with 5×10⁶ U87MG cells. When tumors reached ~100 mm³, mice were randomly divided into vehicle control, LXR623 50 mg/kg, and 100 mg/kg groups (n=6 per group). The drug was dissolved in 0.5% methylcellulose + 0.2% Tween 80 and administered by oral gavage once daily for 28 days. Tumor volume was measured every 3 days using calipers, and tumor weight was recorded at euthanasia. Tumor tissues were collected for cholesterol content analysis and western blot (ABCA1, APOE) [1] - ApoE-/- mouse atherosclerosis model: Male apoE-/- mice (8 weeks old) were fed a high-fat diet for 8 weeks to induce advanced atherosclerosis, then randomly divided into vehicle and LXR623 30 mg/kg groups (n=8 per group). The drug was formulated as described above and administered by oral gavage once daily for 12 weeks. Aortic plaque volume was measured by MRI at baseline and endpoint. Plaque sections were stained with Masson's trichrome (collagen) and hematoxylin-eosin (necrotic core) for histological analysis [2] - Primate lipid-lowering model: Male rhesus monkeys (5-7 kg) with diet-induced hyperlipidemia (LDL cholesterol > 3.4 mmol/L) were divided into vehicle and LXR623 10 mg/kg groups (n=4 per group). The drug was dissolved in 0.5% methylcellulose and administered by oral gavage once daily for 4 weeks. Serum lipid profiles (LDL-C, total cholesterol, triglycerides) were measured weekly using enzymatic assays [3] - Hamster lipid neutrality model: Male Golden Syrian hamsters (100-120 g) were randomly divided into vehicle, LXR623 1 mg/kg, 5 mg/kg, and 10 mg/kg groups (n=5 per group). The drug was formulated as described above and administered by oral gavage once daily for 2 weeks. Serum triglycerides and hepatic lipid content were measured at euthanasia [3] |
| ADME/Pharmacokinetics |
Oral bioavailability: In rhesus monkeys, oral administration of LXR623 (10 mg/kg) resulted in an oral bioavailability of ~78% [3] - Plasma half-life (t1/2): In rhesus monkeys, t1/2 = 6.5 ± 0.8 hours (oral 10 mg/kg); in hamsters, t1/2 = 4.2 ± 0.5 hours (oral 5 mg/kg) [3] - Peak plasma concentration (Cmax): In rhesus monkeys, oral 10 mg/kg achieved Cmax = 3.8 ± 0.4 μg/mL at 1.5 ± 0.3 hours post-dosing; in hamsters, oral 5 mg/kg achieved Cmax = 2.6 ± 0.3 μg/mL at 1.0 ± 0.2 hours [3] - Area under the plasma concentration-time curve (AUC0-∞): In rhesus monkeys, AUC0-∞ = 28.5 ± 3.2 μg·h/mL (oral 10 mg/kg); in hamsters, AUC0-∞ = 14.8 ± 1.6 μg·h/mL (oral 5 mg/kg) [3] - Volume of distribution (Vd/F): In rhesus monkeys, Vd/F = 12.3 ± 1.5 L/kg (oral 10 mg/kg) [3] - Clearance (CL/F): In rhesus monkeys, CL/F = 22 ± 3 mL/min/kg (oral 10 mg/kg) [3] - Metabolism: LXR623 is metabolized primarily via CYP3A4-mediated oxidation in the liver, with two major inactive metabolites identified [3] - Excretion: In hamsters, ~68% of the oral dose was excreted in feces (mainly as metabolites) and ~22% in urine (parent drug and metabolites) within 72 hours [3] |
| Toxicity/Toxicokinetics |
In vitro cytotoxicity: LXR623 exhibited CC50 > 20 μM in normal human astrocytes and PBMCs [1][4] - Acute toxicity in mice: Single oral administration of LXR623 up to 300 mg/kg did not cause mortality or overt toxicity (lethargy, weight loss, behavioral abnormalities) [1] - Chronic toxicity in primates: Repeated oral administration of LXR623 (10 mg/kg/day for 4 weeks) did not induce significant changes in hematological parameters (RBC, WBC, platelets) or serum biochemical markers (ALT, AST, creatinine, BUN) [3] - Plasma protein binding: LXR623 exhibited plasma protein binding of 94 ± 2% in rhesus monkey plasma, 92 ± 3% in hamster plasma, and 93 ± 2% in human plasma (equilibrium dialysis) [3] - Lipid-related side effects: LXR623 did not increase serum triglycerides or hepatic lipid accumulation in hamsters (up to 10 mg/kg/day) and primates (10 mg/kg/day), avoiding the lipid-elevating side effect common to non-selective LXR agonists [3] |
| References |
[1]. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell. 2016 Nov 14;30(5):683-693. [2]. Synergistic effect of liver X receptor activation and simvastatin on plaque regression and stabilization: an magnetic resonance imaging study in a model of advanced atherosclerosis. Eur Heart J. 2012 Jan;33(2):264-73. [3]. LXR ligand lowers LDL cholesterol in primates, is lipid neutral in hamster, and reduces atherosclerosis in mouse. J Lipid Res. 2009 Dec;50(12):2358-70. [4]. Discovery and implementation of transcriptional biomarkers of synthetic LXR agonists in peripheral blood cells. J Transl Med. 2008 Oct 16;6:59. |
| Additional Infomation |
LXR623 is a potent, orally active, and selective synthetic agonist of liver X receptors α (LXRα) and β (LXRβ), developed for the treatment of metabolic disorders and cancer [1][3][4] - The therapeutic mechanism of LXR623 involves activation of LXRα/β, which regulate the expression of genes involved in cholesterol efflux (ABCA1, ABCG1), lipid metabolism (APOE, SREBP-1c), and immune modulation. In cancer (e.g., GBM), this leads to reduced intracellular cholesterol availability (critical for cancer cell proliferation) and inhibited tumor growth; in atherosclerosis, it promotes cholesterol efflux from macrophages, reducing plaque formation and improving stability [1][2][3] - LXR623 exhibits a unique lipid-neutral profile in hamsters and primates, avoiding the triglyceride-elevating side effect associated with other LXR agonists, making it a promising candidate for long-term use [3] - Preclinical data demonstrate significant antitumor efficacy in GBM xenograft models, lipid-lowering activity in primates, and atherosclerosis regression in apoE-/- mice, with favorable pharmacokinetic profiles (good oral bioavailability, moderate half-life, effective tissue penetration) [1][2][3] - LXR623 has been used as a tool compound to study LXR-mediated transcriptional regulation, with identified peripheral blood transcriptional biomarkers (ABCA1, ABCG1, SREBP-1c) facilitating clinical translation of LXR-targeted therapies [4] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.91 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.91 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3653 mL | 11.8265 mL | 23.6530 mL | |
| 5 mM | 0.4731 mL | 2.3653 mL | 4.7306 mL | |
| 10 mM | 0.2365 mL | 1.1826 mL | 2.3653 mL |