Physicochemical Properties
| Molecular Formula | C7H15CL2N2O2P |
| Molecular Weight | 261.08 |
| Exact Mass | 260.025 |
| CAS # | 66849-34-1 |
| PubChem CID | 3690 |
| Appearance |
Crystals from anhyd ether White crystalline powder |
| Density | 1.334g/cm3 |
| Boiling Point | 336.099ºC at 760 mmHg |
| Melting Point |
39-41 °C 39-41 °C 39 - 41 °C |
| Flash Point | 157.068ºC |
| Index of Refraction | 1.506 |
| LogP | 2.212 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 4 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 14 |
| Complexity | 218 |
| Defined Atom Stereocenter Count | 0 |
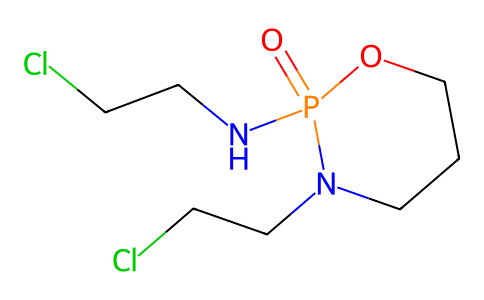
| SMILES | C1CN(P(=O)(OC1)NCCCl)CCCl |
| InChi Key | HOMGKSMUEGBAAB-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C7H15Cl2N2O2P/c8-2-4-10-14(12)11(6-3-9)5-1-7-13-14/h1-7H2,(H,10,12) |
| Chemical Name | N,3-bis(2-chloroethyl)-2-oxo-1,3,2λ5-oxazaphosphinan-2-amine |
| Synonyms | L-Ifosfamide; (+)-Ifosfamide |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Ifosfamide is extensively metabolized in humans and the metabolic pathways appear to be saturated at high doses. After administration of doses of 5 g/m2 of 14C-labeled ifosfamide, from 70% to 86% of the dosed radioactivity was recovered in the urine, with about 61% of the dose excreted as parent compound. At doses of 1.6–2.4 g/m2 only 12% to 18% of the dose was excreted in the urine as unchanged drug within 72 hours. Ifosfamide volume of distribution (Vd) approximates the total body water volume, suggesting that distribution takes place with minimal tissue binding. Following intravenous administration of 1.5 g/m2 over 0.5 hour once daily for 5 days to 15 patients with neoplastic disease, the median Vd of ifosfamide was 0.64 L/kg on Day 1 and 0.72 L/kg on Day 5. When given to pediatric patients, the volume of distribution was 21±1.6 L/m^2. 2.4±0.33 L/h/m^2 [pediatric patients] Renal excretion & t1/2 are dose & schedule dependent. 60-80% recovered as unchanged drug or metabolite in urine within 72 hr after admin. The distribution of ifosfamide (IF) and its metabolites 2-dechloroethylifosfamide (2DCE), 3-dechloroethylifosfamide (3DCE), 4-hydroxyifosfamide (4OHIF) and ifosforamide mustard (IFM) between plasma and erythrocytes was examined in vitro and in vivo. In vitro distribution was investigated by incubating blood with various concentrations of IF and its metabolites. In vivo distribution of IF, 2DCE, 3DCE and 4OHIF was determined in 7 patients receiving 9 g/m(2)/72 h intravenous continuous IF infusion. In vitro distribution equilibrium between erythrocytes and plasma was obtained quickly after drug addition. Mean (+/-sem) in vitro and in vivo erythrocyte (e)-plasma (p) partition coefficients (P(e/p)) were 0.75+/-0.01 and 0.81+/-0.03, 0.62+/-0.09 and 0.73+/-0.05, 0.76+/-0.10 and 0.93+/-0.05 and 1.38+/-0.04 and 0.98+/-0.09 for IF, 2DCE, 3DCE and 4OHIF, respectively. These ratios were independent of concentration and unaltered with time. The ratios of the area under the erythrocyte and plasma concentration--time curves (AUC(e/p)) were 0.96+/-0.03, 0.87+/-0.07, 0.98+/-0.06 and 1.34+/-0.39, respectively. A time- and concentration-dependent distribution--equilibrium phenomenon was observed with the relative hydrophilic IFM. It is concluded that IF and metabolites rapidly reach distribution equilibrium between erythrocytes and plasma; the process is slower for IFM. Drug distribution to the erythrocyte fraction ranged from about 38% for 2DCE to 58% for 4OHIF, and was stable over a wide range of clinically relevant concentrations. A strong parallelism in the erythrocyte and plasma concentration profiles was observed for all compounds. Thus, pharmacokinetic assessment using only plasma sampling yields direct and accurate insights into the whole blood kinetics of IF and metabolites and may be used for pharmacokinetic-pharmacodynamic studies. ... To assess the feasibility of a sparse sampling approach for the determination of the population pharmacokinetics of ifosfamide, 2- and 3-dechloroethyl-ifosfamide and 4-hydroxy-ifosfamide in children treated with single-agent ifosfamide against various malignant tumours. ... Pharmacokinetic assessment followed by model fitting. Patients: The analysis included 32 patients aged between 1 and 18 years receiving a total of 45 courses of ifosfamide 1.2, 2 or 3 g/m2 in 1 or 3 hours on 1, 2 or 3 days. ... A total of 133 blood samples (median of 3 per patient) were collected. Plasma concentrations of ifosfamide and its dechloroethylated metabolites were determined by gas chromatography. Plasma concentrations of 4-hydroxy-ifosfamide were measured by high-performance liquid chromatography. The models were fitted to the data using a nonlinear mixed effects model as implemented in the NONMEM program. A cross-validation was performed. ... Population values (mean +/- standard error) for the initial clearance and volume of distribution of ifosfamide were estimated at 2.36 +/- 0.33 L/h/m2 and 20.6 +/- 1.6 L/m2 with an interindividual variability of 43 and 32%, respectively. The enzyme induction constant was estimated at 0.0493 +/- 0.0104 L/h2/m2. The ratio of the fraction of ifosfamide metabolised to each metabolite to the volume of distribution of that metabolite, and the elimination rate constant, of 2- and 3-dechloroethyl-ifosfamide and 4-hydroxy-ifosfamide were 0.0976 +/- 0.0556, 0.0328 +/- 0.0102 and 0.0230 +/- 0.0083 m2/L and 3.64 +/- 2.04, 0.445 +/- 0.174 and 7.67 +/- 2.87 h(-1), respectively. Interindividual variability of the first parameter was 23, 34 and 53%, respectively. Cross-validation indicated no bias and minor imprecision (12.5 +/- 5.1%) for 4-hydroxy-ifosfamide only. ... We have developed and validated a model to estimate ifosfamide and metabolite concentrations in a paediatric population by using sparse sampling. ... The population pharmacokinetics and pharmacodynamics of the cytostatic agent ifosfamide and its main metabolites 2- and 3-dechloroethylifosfamide and 4-hydroxyifosfamide were assessed in patients with soft tissue sarcoma. ... Twenty patients received 9 or 12 g/m2 ifosfamide administered as a 72-h continuous intravenous infusion. The population pharmacokinetic model was built in a sequential manner, starting with a covariate-free model and progressing to a covariate model with the aid of generalised additive modelling. ... The addition of the covariates weight, body surface area, albumin, serum creatinine, serum urea, alkaline phosphatase and lactate dehydrogenase improved the prediction errors of the model. Typical pretreatment (mean +/- SEM) initial clearance of ifosfamide was 3.03 +/- 0.18 l/h with a volume of distribution of 44.0 +/- 1.8 l. Autoinduction, dependent on ifosfamide levels, was characterised by an induction half-life of 11.5 +/- 1.0 h with 50% maximum induction at 33.0 +/- 3.6 microM ifosfamide. Significant pharmacokinetic-pharmacodynamic relationships (P = 0.019) were observed between the exposure to 2- and 3-dechloroethylifosfamide and orientational disorder, a neurotoxic side-effect. No pharmacokinetic-pharmacodynamic relationships between exposure to 4-hydroxyifosfamide and haematological toxicities could be observed in this population. For more Absorption, Distribution and Excretion (Complete) data for IFOSFAMIDE (6 total), please visit the HSDB record page. Metabolism / Metabolites Primarily hepatic. Ifosfamide is metabolized through two metabolic pathways: ring oxidation ("activation") to form the active metabolite, 4-hydroxy-ifosfamide and side-chain oxidation to form the inactive metabolites, 3-dechloro-ethylifosfamide or 2-dechloroethylifosfamide with liberation of the toxic metabolite, chloroacetaldehyde. Small quantities (nmol/mL) of ifosfamide mustard and 4-hydroxyifosfamide are detectable in human plasma. Metabolism of ifosfamide is required for the generation of the biologically active species and while metabolism is extensive, it is also quite variable among patients. Like cyclophosphamide, ifosfamide is activated in the liver by hydroxylation. However, the activation of ifosfamide proceeds more slowly, with greater production of dechlorinated metabolites & chloroacetaldehyde. These differences in metabolism likely account for the higher doses of ifosfamide required for equitoxic effects & the possible difference in antitumor spectrum of the two agents. Like cyclophosphamide, isophosphamide requires metabolism by microsomal enzymes to act as a cytotoxic agent. It is rapidly metabolized in many species, including rodents and dogs; the urinary metabolites indicate that a series of reactions take place analogous to those in the metabolism of cyclophosphamide. Acrolein is produced during its oxidative degradation, and one product of the reaction is the ring-opened carboxy derivative. Dogs also rapidly metabolize isophosphamide, and the carboxy derivative and 4-keto isophosphamide have been identified in the urine. The aim of this study was to develop a population pharmacokinetic model that could describe the pharmacokinetics of ifosfamide. 2- and 3-dechloroethylifosfamide and 4-hydroxyifosfamide, and calculate their plasma exposure and urinary excretion. A group of 14 patients with small-cell lung cancer received a 1-h intravenous infusion of 2.0 or 3.0 g/m2 ifosfamide over 1 or 2 days in combination with 175 mg/m2 paclitaxel and carboplatin at AUC 6. The concentration-time profiles of ifosfamide were described by an ifosfamide concentration-dependent development of autoinduction of ifosfamide clearance. Metabolite compartments were linked to the ifosfamide compartment enabling description of the concentration-time profiles of 2- and 3-dechloroethylifosfamide and 4-hydroxyifosfamide. The Bayesian estimates of the pharmacokinetic parameters were used to calculate the systemic exposure to ifosfamide and its metabolites for the four ifosfamide schedules. Fractionation of the dose over 2 days resulted increased metabolite formation, especially of 2-dechloroethylifosfamide, probably due to increased autoinduction. Renal recovery was only minor with 6.6% of the administered dose excreted unchanged and 9.8% as dechloroethylated metabolites. In conclusion, ifosfamide pharmacokinetics were described with an ifosfamide concentration-dependent development of autoinduction and allowed estimation of the population pharmacokinetics of the metabolites of ifosfamide. Fractionation of the dose resulted in increased exposure to 2-dechloroethylifosfamide, probably due to increased autoinduction. The anticancer drug ifosfamide is a prodrug requiring activation through 4-hydroxyifosfamide to ifosforamide mustard, to exert cytotoxicity. Deactivation of ifosfamide leads to 2- and 3-dechloroethylifosfamide and the release of potentially neurotoxic chloracetaldehyde. The aim of this study was to quantify and to compare the pharmacokinetics of ifosfamide, 2- and 3-dechloroethylifosfamide, 4-hydroxyifosfamide, and ifosforamide mustard in short (1-4 h), medium (24-72 h), and long infusion durations (96-240 h) of ifosfamide. An integrated population pharmacokinetic model was used to describe the autoinducible pharmacokinetics of ifosfamide and its four metabolites in 56 patients. The rate by which autoinduction of the metabolism of ifosfamide developed was found to be significantly dependent on the infusion schedule. The rate was 52% lower with long infusion durations compared with short infusion durations. This difference was, however, comparable with its interindividual variability (22%) and was, therefore, considered to be of minor clinical importance. Autoinduction caused a less than proportional increase in the area under the ifosfamide plasma concentration-time curve (AUC) and more than proportional increase in metabolite exposure with increasing ifosfamide dose. During long infusion durations dose-corrected exposures (AUC/D) were significantly decreased for ifosfamide and increased for 3-dechloroethylifosfamide compared with short infusion durations. No differences in dose-normalized exposure to ifosfamide and metabolites were observed between short and medium infusion durations. This study demonstrates that the duration of ifosfamide infusion influences the exposure to the parent and its metabolite 3-dechloroethylifosfamide. The observed dose and infusion duration dependence should be taken into account when modeling ifosfamide metabolism. Ifosfamide is a known human metabolite of L-trofosfamide. Primarily hepatic. Ifosfamide is metabolized through two metabolic pathways: ring oxidation ("activation") to form the active metabolite, 4-hydroxy-ifosfamide and side-chain oxidation to form the inactive metabolites, 3-dechloro-ethylifosfamide or 2-dechloroethylifosfamide with liberation of the toxic metabolite, chloroacetaldehyde. Small quantities (nmol/mL) of ifosfamide mustard and 4-hydroxyifosfamide are detectable in human plasma. Metabolism of ifosfamide is required for the generation of the biologically active species and while metabolism is extensive, it is also quite variable among patients. Route of Elimination: Ifosfamide is extensively metabolized in humans and the metabolic pathways appear to be saturated at high doses. After administration of doses of 5 g/m2 of 14C-labeled ifosfamide, from 70% to 86% of the dosed radioactivity was recovered in the urine, with about 61% of the dose excreted as parent compound. At doses of 1.6 - 2.4 g/m2 only 12% to 18% of the dose was excreted in the urine as unchanged drug within 72 hours. Half Life: 7-15 hours. The elimination half-life increase appeared to be related to the increase in ifosfamide volume of distribution with age. Biological Half-Life 7-15 hours. The elimination half-life increase appeared to be related to the increase in ifosfamide volume of distribution with age. The elimination half-life associated with doses of 2.5 g/sq m is 6-8 hr, whereas the elimination half-life associated with doses of 3.5-5 g/sq m is 14-16 hr. |
| Toxicity/Toxicokinetics |
Toxicity Summary After metabolic activation, active metabolites of ifosfamide alkylate or bind with many intracellular molecular structures, including nucleic acids. The cytotoxic action is primarily due to cross-linking of strands of DNA and RNA, as well as inhibition of protein synthesis. Hepatotoxicity The toxicity of ifosfamide seems to be similar to that of cyclophosphamide. Mild and transient elevations in serum aminotransferase levels are found in a high proportion of patients receiving ifosfamide. Because ifosfamide is typically given in combination with other antineoplastic agents, its role in causing the serum enzyme elevations is often not clear. The abnormalities are generally transient, do not cause symptoms and do not require dose modification. Clinically apparent liver injury from ifosfamide has been limited to a small number of cases of cholestatic hepatitis arising within a few weeks of receiving ifosfamide (with other antineoplastic agents). In addition, sinusoidal obstruction syndrome has been reported after conditioning regimens that have included ifosfamide in preparation for hematopoietic cell transplantation. The onset of injury is usually within one to three weeks of the myeloablation and is characterized by a sudden onset of abdominal pain, weight gain, ascites, marked increase in serum aminotransferase levels (and lactic dehydrogenase), and subsequent jaundice and hepatic dysfunction. The severity of sinusoidal obstruction syndrome varies from a transient, self limited injury to acute liver failure. The diagnosis is usually based on clinical features of tenderness and enlargement of the liver, weight gain, ascites and jaundice. Liver biopsy is diagnostic but often contraindicated, because of severe thrombocytopenia after bone marrow transplantation. Likelihood score: D (possible rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Most sources consider breastfeeding to be contraindicated during maternal antineoplastic drug therapy, especially alkylating agents such as ifosfamide. Labeling suggests that mothers should not breastfeed during therapy and for 1 week after the last dose of ifosfamide or mesna. Chemotherapy may adversely affect the normal microbiome and chemical makeup of breastmilk. Women who receive chemotherapy during pregnancy are more likely to have difficulty nursing their infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Ifosfamide shows little plasma protein binding. Toxicity Data LD50 (mouse) = 390-1005 mg/kg, LD50 (rat) = 150-190 mg/kg. Interactions The more urotoxic agent ifosfamide was introduced to the market with the uroprotective agent, Mesna. Mesna liberated free thiol groups in the bladder which then can react with & neutralize the oxazaphosphorine metabolite. When administered in an appropriate dosing schedule, Mesna can prevent the bladder toxicity completely. BACKGROUND: The autoinducible metabolic transformation of the anticancer agent ifosfamide involves activation through 4-hydroxyifosfamide to the ultimate cytotoxic ifosforamide mustard and deactivation to 2- and 3-dechloroethylifosfamide with concomitant release of the neurotoxic chloroacetaldehyde. Activation is mediated by cytochrome P450 (CYP) 3A4 and deactivation by CYP3A4 and CYP2B6. The aim of this study was to investigate modulation of the CYP-mediated metabolism of ifosfamide with ketoconazole, a potent inhibitor of CYP3A4, and rifampin (INN, rifampicin), an inducer of CYP3A4/CYP2B6. METHODS: In a double-randomized, 2-way crossover study a total of 16 patients received ifosfamide 3 g/m(2) per 24 hours intravenously, either alone or in combination with 200 mg ketoconazole twice daily (1 day before treatment and 3 days of concomitant administration) or 300 mg rifampin twice daily (3 days before treatment and 3 days of concomitant administration). Plasma pharmacokinetics and urinary excretion of ifosfamide, 2- and 3-dechloroethylifosfamide, and 4-hydroxyifosfamide were assessed in both courses. Data analysis was performed with a population pharmacokinetic model with a description of autoinduction of ifosfamide. RESULTS: Rifampin increased the clearance of ifosfamide at the start of therapy at 102%. The fraction of ifosfamide metabolized to the dechloroethylated metabolites was increased, whereas exposure to the metabolites was decreased as a result of increased elimination. The fraction metabolized and the exposure to 4-hydroxyifosfamide were not significantly influenced. Ketoconazole did not affect the fraction metabolized or the exposure to the dechloroethylated metabolites, whereas both parameters were reduced with 4-hydroxyifosfamide. CONCLUSIONS: Coadministration of ifosfamide with ketoconazole or rifampin did not produce changes in the pharmacokinetics of the parent or metabolites that may result in an increased benefit of ifosfamide therapy. Leukopenic and/or thrombocytopenic effects of ifosfamide may be increased with concurrent or recent therapy if these medications /blood dyscarasia-causing medications/ cause the same effects; dosage adjustments of ifosfamide, if necessary, should be based on blood counts. Additive bone marrow depression may occur; dosage reduction may be required when two or more bone marrow depressants, including radiation, are used concurrently or consecutively /with ifosfamide/. For more Interactions (Complete) data for IFOSFAMIDE (7 total), please visit the HSDB record page. Non-Human Toxicity Values LD50 Rat oral 143 mg/kg LD50 Rat ip 140 mg/kg LD50 Rat sc 160 mg/kg LD50 Rat iv 190 mg/kg For more Non-Human Toxicity Values (Complete) data for IFOSFAMIDE (8 total), please visit the HSDB record page. |
| Additional Infomation |
Therapeutic Uses Ifosfamide currently is approved for use in combination with other drugs for germ cell testicular cancer & is widely used to treat pediatric & adult sarcomas. Clinical trials also have shown ifosfamide to be active against carcinomas of the cervix & lung & against lymphomas. It is a common component of high-dose chemotherapy regimens with bone marrow or stem cell rescue; in these regimens, in total doses of 12-14 g/sq m, it may cause severe neurological toxicity, including coma & death. This toxicity is thought to result form a metabolite, chloracetaldehyde. In addition to hemorrhagic cystitis, ifosfamide causes nausea, vomiting, anorexia, leukopenia, nephrotoxicity, & CNS disturbances (especially somnolence & confusion). Ifosfamide is indicated, in combination with other antineoplastic agents and a prophylactic agent against hemorrhagic cystitis (such as mesna), for treatment of germ cell testicular tumors. /Included in US product labeling/ Ifosfamide is indicated as reasonable medical therapy for treatment of head and neck carcinoma. (Evidence rating: IIID) /NOT included in US product labeling/ Ifosfamide is used for treatment of soft-tissue sarcomas, Ewing's sarcoma, and Hodgkin's and non-Hodgkin's lymphomas. /NOT included in US product labeling/ For more Therapeutic Uses (Complete) data for IFOSFAMIDE (9 total), please visit the HSDB record page. Drug Warnings It is a common component of high-dose chemotherapy regimens with bone marrow or stem cell rescue; in these regimens, in total doses of 12-14 g/sq m, it may cause severe neurological toxicity, including coma & death. This toxicity is thought to result form a metabolite, chloracetaldehyde. In addition to hemorrhagic cystitis, ifosfamide causes nausea, vomiting, anorexia, leukopenia, nephrotoxicity, & CNS disturbances (especially somnolence & confusion). Ifosfamide is distributed into breast milk. Breast feeding is not recommended during chemotherapy because of the risks to the infant (adverse effects, mutagenicity, carcinogenicity). The bone marrow depressant effects of ifosfamide may result in an increased incidence of microbial infection, delayed healing, and gingival bleeding. Dental work, whenever possible, should be completed prior to initiation of therapy or deferred until blood counts have returned to normal. Patients should be instructed in proper oral hygiene during treatment, including caution in use of regular toothbrushes, dental floss, and toothpicks. Many side effects of antineoplastic therapy are unavoidable and represent the medication's pharmacologic action. Some of these (for example, leukopenia and thrombocytopenia) are actually used as parameters to aid in individual dosage titration. For more Drug Warnings (Complete) data for IFOSFAMIDE (20 total), please visit the HSDB record page. Pharmacodynamics Ifosfamide requires activation by microsomal liver enzymes to active metabolites in order to exert its cytotoxic effects. Activation occurs by hydroxylation at the ring carbon atom 4 to form the unstable intermediate 4-hydroxyifosfamide. This metabolite than rapidly degrades to the stable urinary metabolite 4-ketoifosfamide. The stable urinary metabolite, 4-carboxyifosfamide, is formed upon opening of the ring. These urinary metabolites have not been found to be cytotoxic. N, N-bis (2-chloroethyl)-phosphoric acid diamide (ifosphoramide) and acrolein are also found. The major urinary metabolites, dechloroethyl ifosfamide and dechloroethyl cyclophosphamide, are formed upon enzymatic oxidation of the chloroethyl side chains and subsequent dealkylation. It is the alkylated metabolites of ifosfamide that have been shown to interact with DNA. Ifosfamide is cycle-phase nonspecific. |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.8302 mL | 19.1512 mL | 38.3024 mL | |
| 5 mM | 0.7660 mL | 3.8302 mL | 7.6605 mL | |
| 10 mM | 0.3830 mL | 1.9151 mL | 3.8302 mL |