KU-55933 is a potent and specific ATM (Ataxia-telangiectasia mutated) kinase inhibitor with IC50/Ki of 12.9 nM/2.2 nM in cell-free assays, and is highly selective for ATM as compared to DNA-PK, PI3K/PI4K, ATR and mTOR. As an ATM inhibitor, KU-55933 dramatically reduced the rise in phospho-Akt at Ser473 in insulin- and IGF-I-treated MDA-MB-453 and PC-3 cells after serum starvation. In MDA-MB-453 and PC-3 cells, KU-55933 treatment reduced cell proliferation in the MTT assay by roughly 50% at a concentration of 10 μM. Treatment with KU-55933 inhibited cell proliferation in a panel of cell lines with varying Akt activities, and this effect was correlated with Akt phosphorylation. This new understanding of the mechanism governing ATM regulation may be helpful in developing more accurate plans for modulating ATM activity in cancer therapy, since it is thought that ATM inhibition makes cancer cells more susceptible to genotoxic substances.
Physicochemical Properties
| Molecular Formula | C21H17NO3S2 | |
| Molecular Weight | 395.49 | |
| Exact Mass | 395.064 | |
| Elemental Analysis | C, 63.77; H, 4.33; N, 3.54; O, 12.14; S, 16.22 | |
| CAS # | 587871-26-9 | |
| Related CAS # |
|
|
| PubChem CID | 5278396 | |
| Appearance | White to off-white solid powder | |
| Density | 1.4±0.1 g/cm3 | |
| Boiling Point | 628.0±55.0 °C at 760 mmHg | |
| Melting Point | 229.98° C | |
| Flash Point | 333.6±31.5 °C | |
| Vapour Pressure | 0.0±1.8 mmHg at 25°C | |
| Index of Refraction | 1.714 | |
| LogP | 6.13 | |
| Hydrogen Bond Donor Count | 0 | |
| Hydrogen Bond Acceptor Count | 6 | |
| Rotatable Bond Count | 2 | |
| Heavy Atom Count | 27 | |
| Complexity | 643 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | S1C2=C([H])C([H])=C([H])C([H])=C2SC2=C([H])C([H])=C([H])C(=C12)C1=C([H])C(C([H])=C(N2C([H])([H])C([H])([H])OC([H])([H])C2([H])[H])O1)=O |
|
| InChi Key | XRKYMMUGXMWDAO-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C21H17NO3S2/c23-14-12-16(25-20(13-14)22-8-10-24-11-9-22)15-4-3-7-19-21(15)27-18-6-2-1-5-17(18)26-19/h1-7,12-13H,8-11H2 | |
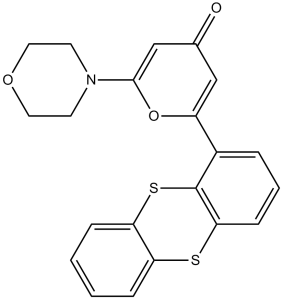
| Chemical Name | 2-morpholin-4-yl-6-thianthren-1-ylpyran-4-one | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | ATM ( IC50 = 12.9 nM ); DNA-PK ( IC50 = 2500 nM ); mTOR ( IC50 = 9300 nM ); PI3K ( IC50 = 16600 nM ) | |
| ln Vitro |
|
|
| ln Vivo |
ATM kinase ablation ameliorates cell cycle disorders and insults of podocytes in mice[4] To further confirm the role of ATM in ADR-related podocyte cell cycle reentry, researchers used a specific ATM kinase inhibitor, KU-55933, to suppress the phosphorylation and activity of ATM. Strikingly, KU-55933 alleviated MAD2B elevation (P<0.05) caused by ADR stimulation (Figure 6A-B). Simultaneously, the alteration of Skp2 and p27, the well-known substrates of MAD2B and pivotal regulators of cell cycle, induced by ADR was partially reversed (P<0.05) by KU-55933 (Figure 6C-D). Moreover, flow cytometric analysis revealed that the ATM inhibitor increased podocytes arrested in G2/M-phase (P<0.05) (Figure 6E-F), avoiding disastrous division and following cell death. In addition, KU-55933 successfully prevented ADR-triggered podocyte dysfunction, as indicated by the recovery of nephrin (P<0.05) and CD2AP (P<0.05) expression (Figure 6G-H). Consistently, KU-55933 effectively suppressed the overexpression of MAD2B (P<0.05) provoking by ADR injection in mice (Figure 7A-B). The morphological abnormalities of FSGS were minimized by pretreatment with KU-55933 (Figure 7C-E), along with lower proteinuria (P<0.05) and elevated serum albumin (P<0.05) (Figure 7F-G). Therefore, our observation suggests that blocking ATM activation could effectively prevent or mitigate podocyte injury. |
|
| Enzyme Assay | In order to obtain ATM for the in vitro assay, rabbit polyclonal antiserum raised to the COOH-terminal 400 amino acids of ATM is immunoprecipitated from HeLa nuclear extract using a method that involves buffering the mixture with 25 mM HEPES (pH 7.4), 2 mM MgCl2, 250 mM KCl, 500 μM EDTA, 100 μM Na3VO4, 10% v/v glycerol, and 0.1% v/v Igepal. After an hour of incubation with protein A-Sepharose beads and subsequent centrifugation to recover the beads, ATM-antibody complexes are separated from nuclear extract. A 96-well plate's well is used to incubate ATM-containing Sepharose beads with 1 μg of glutathione S-transferase–p53N66 (p53's NH2-terminal 66 amino acids fused to glutathione S-transferase) in the ATM assay buffer [25 mM HEPES (pH 7.4), 75 mM NaCl, 3 mM MgCl2, 2 mM MnCl2, 50 μM Na3VO4, 500 μM DTT, and 5% v/v glycerol] at 37 °C with or without an inhibitor. The reaction is continued at 37 °C for an additional hour after adding ATP to a final concentration of 50 μM after 10 minutes of gentle shaking. Glutathione S-transferase-p53N66 binding is allowed to occur by centrifuging the plate at 250 × g for 10 minutes (4 °C) in order to remove the beads containing ATM. The supernatant is then taken out and put in a white opaque 96-well plate. This incubation process takes 1.5 hours at room temperature. The PBS wash, dry blotting, and standard ELISA analysis using a phospho-serine 15 p53 antibody are the next steps for this plate. When using a secondary antibody conjugated with horseradish peroxidase from goat antimouse, the substrate for phosphorylated glutathione S-transferase-p53N66 is detected. The process of creating a signal and chemiluminescent detection involves using an enhanced chemiluminescence solution. Chemiluminescent detection is done and a signal is generated using an enhanced chemiluminescence solution. | |
| Cell Assay | The ATM response is measured by Western blot analysis of p53 serine 15 phosphorylation and stabilization of wild-type p53 in U2OS cells that have been exposed to ionizing radiation (3, 5, or 15 Gy) or UV (5 or 50 J/m2). Each time point's whole cell extracts are extracted, proteins are separated using SDS-PAGE, and a p53 phospho-serine 15 specific antibody is used to measure the ATM-specific increase in phosphorylated serine 15. When using a p53-specific antibody (DO-1), overall p53 stabilization over time is also seen. Similarly, the following antibodies are used to study ATM-dependent phosphorylations on H2AX, CHK1, NBS1, and SMC1: NBS1 phospho-serine 343 and CHK1 phospho-serine 345 antibodies. SMC1 and SMC1 phospho-serine 966 antibodies are also used, along with antibodies against histone H2A (H-124) and CHK1. The peak response time of two hours for p53 serine 15 phosphorylation is used to track ATM inhibition in order to determine a cellular IC50 for KU-55933. Prior to applying ionizing radiation, KU-55933 is titrated onto cells and preincubated for one hour. The IC50 value is determined similarly to the in vitro determinations, and the percentage inhibition in relation to the vehicle control is computed using scanning densitometry. | |
| Animal Protocol |
ADR-induced FSGS murine model[4] Adult male mice (8 wks of age with Balb/C background) weighing 21-24 g were raised in a specific pathogen-free environment with a 12 h light/dark cycle, and allowed access to food and water ad libitum. To establish the FSGS animal model, the mice were injected with a single dose of ADR (15 mg/kg) via the tail vein and sacrificed after 4 wks. Urine and serum samples were harvested prior to sacrifice, and the urine protein/creatinine, serum albumin, creatinine, and blood urea nitrogen were measured using an automated chemistry analyzer. After flushing with ice-cold Krebs-Henseleit-saline buffer via an aortal catheter, the kidneys were dissected on ice. The cortex tissues were snap frozen and stored at -80°C until use. To inhibit ATM kinase, KU-55933 (500 µg/kg), a specific ATM inhibitor, was dissolved in 0.15% DMSO and was administered intraperitoneally 24 h prior to ADR injection and repeated every 3 days until sacrifice.[4] BALB/c nu/nu nude mice bearing LU1205 cells 10 μM |
|
| References |
[1]. Discovery of 2-[1-(4,4-Difluorocyclohexyl)piperidin-4-yl]-6-fluoro-3-oxo-2,3-dihydro-1H-isoindole-4-carboxamide (NMS-P118): A Potent, Orally Available, and Highly Selective PARP-1 Inhibitor for Cancer Therapy. J Med Chem. 2015 Sep 10;58(17):6875-98. [2]. Mechanisms of chemotherapy-induced human ovarian aging: double strand DNA breaks and microvascular compromise. Aging (Albany NY). 2011 Aug;3(8):782-93. [3]. Inhibition of ataxia telangiectasia mutated kinase activity enhances TRAIL-mediated apoptosis in human melanoma cells. Cancer Res. 2009 Apr 15;69(8):3510-9. [4]. MAD2B-mediated cell cycle reentry of podocytes is involved in the pathogenesis of FSGS. Int J Biol Sci . 2021 Oct 22;17(15):4396-4408. |
|
| Additional Infomation |
ATM Kinase Inhibitor is any agent that inhibits ataxia telangiectasia mutated (ATM) kinase. The mechanism of chemotherapy-induced acceleration of ovarian aging is not fully understood. We used doxorubicin, a widely used cancer chemotherapeutic, in a variety of in vivo xenograft, and in vitro models to investigate the impact of chemotherapy-induced aging on the human ovary. Doxorubicin caused massive double-strand-DNA-breaks in primordial follicles, oocytes, and granulosa cells in a dose dependent fashion as revealed by accumulating γH2AX foci. This damage was associated with apoptotic oocyte death and resulted in the activation of ATM. It appeared that the repair response enabled a minor proportion of oocytes (34.7%) and granulosa cells (12.1%) to survive while the majority succumbed to apoptotic death. Paradoxically, inhibition of ATM by KU-55933 resulted in improved survival, probably via prevention of downstream activation of TAp63α. Furthermore, doxorubicin caused vascular and stromal damage in the human ovary, which might impair ovarian function both pre- and post-menopausally. Chemotherapy-induced premature ovarian aging appears to result from a complex process involving both the germ- and non-germ cell components of the ovary. These effects may have clinical implications in aging both for premenopausal and postmenopausal cancer survivors.[2] The aim of the present study was to elucidate the effects of ataxia telangiectasia mutated (ATM) kinase on the regulation of the extrinsic tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor 2/DR5-mediated death pathway in human melanoma cells. We revealed that total ATM protein levels were high in some human melanoma lines compared with normal cells. The basal levels of active form ATM phospho-Ser(1981) were also detectable in many melanoma lines and could be further up-regulated by gamma-irradiation. Pretreatment of several melanoma lines just before gamma-irradiation with the inhibitor of ATM kinase KU-55933 suppressed p53 and nuclear factor-kappaB (NF-kappaB) activation but notably increased radiation-induced DR5 surface expression, down-regulated cFLIP (caspase-8 inhibitor) levels, and substantially enhanced exogenous TRAIL-induced apoptosis. Furthermore, gamma-irradiation in the presence of KU-55933 rendered TRAIL-resistant HHMSX melanoma cells susceptible to TRAIL-mediated apoptosis. In addition, suppression of ATM expression by the specific short hairpin RNA also resulted in down-regulation of cFLIP levels, up-regulation of surface DR5 expression, and TRAIL-mediated apoptosis in melanoma cells. Besides p53 and NF-kappaB, crucial regulators of DR5 expression, transcription factor STAT3 is known to negatively regulate DR5 expression. Suppression of Ser(727) and Tyr(705) phosphorylation of STAT3 by KU-55933 reduced STAT3 transacting activity accompanied by elevation in DR5 expression. Dominant-negative STAT3beta also efficiently up-regulated the DR5 surface expression and down-regulated cFLIP levels in melanoma cells in culture and in vivo. Taken together, our data show the existence of an ATM-dependent STAT3-mediated antiapoptotic pathway, which on suppression sensitizes human melanoma cells to TRAIL-mediated apoptosis.[3] Rationale: Focal segmental glomerulosclerosis (FSGS) is characterized by the dysfunction of "post-mitotic" podocytes. The reentry of podocytes in the cell cycle will ultimately result in cell death. Mitotic arrest deficient 2-like protein 2 (MAD2B), an inhibitor of anaphase-promoting complex (APC)/cyclosome, precisely controls the metaphase to anaphase transition and ordered cell cycle progression. However, the role of MAD2B in FSGS podocyte injury remains unknown. Methods: To explore MAD2B function in podocyte cell cycle reentry, we used conditional mutant mice lacking MAD2B selectively in podocytes in ADR-induced FSGS murine model. Additionally, KU-55933, a specific inhibitor of ataxia-telangiectasia mutated (ATM) was utilized in vivo and in vitro to explore the role of ATM in regulating MAD2B. Results: The expression of MAD2B in podocytes was dramatically increased in patients with FSGS and ADR-treated mice along with podocyte cell cycle reentry. Podocyte-specific knockout of MAD2B effectively attenuated proteinuria, podocyte injury, and prevented the aberrant cell cycle reentry. By bioinformatics analysis we revealed that ATM kinase is a key upstream regulator of MAD2B. Furthermore, inhibition of ATM kinase abolished MAD2B-driven cell cycle reentry and alleviated podocyte impairment in FSGS murine model. In vitro studies by site-directed mutagenesis and immunoprecipitation we revealed ATM phosphorylated MAD2B and consequently hampered the ubiquitination of MAD2B in a phosphorylation-dependent manner. Conclusions: ATM kinase-MAD2B axis importantly contributes to the cell cycle reentry of podocytes, which is a novel pathogenic mechanism of FSGS, and may shed light on the development of its therapeutic approaches.[4] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (6.32 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (6.32 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (6.32 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 5% DMSO and 47.5% PEG300: 10mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.5285 mL | 12.6425 mL | 25.2851 mL | |
| 5 mM | 0.5057 mL | 2.5285 mL | 5.0570 mL | |
| 10 mM | 0.2529 mL | 1.2643 mL | 2.5285 mL |