KPT-185 (KPT185; KPT 185) is an irreversible and selective CRM1 (Chromosomal Maintenance 1, also known as Exportin 1 or XPO1) inhibitor with potential antitumor activity. In AML cell lines, KPT-185 inhibits proliferation of a variety of leukemia cells with IC50 values ranging from 100nM to 500nM. It induces cell-cycle arrest at G1 and induces apoptosis. KPT-185 also strongly affects cell colony formation. In addition, the inhibition of CRM1 caused by KPT-185 induces differentiation of AML blast. Besides that, KPT-185 is also found to inhibit proliferation and induce apoptosis of pancreatic cancer cells including Colo-357, HPAC and BxPC-3.
Physicochemical Properties
| Molecular Formula | C16H16F3N3O3 | |
| Molecular Weight | 355.31 | |
| Exact Mass | 355.114 | |
| Elemental Analysis | C, 54.09; H, 4.54; F, 16.04; N, 11.83; O, 13.51 | |
| CAS # | 1333151-73-7 | |
| Related CAS # |
|
|
| PubChem CID | 53495165 | |
| Appearance | White to off-white solid powder | |
| Density | 1.3±0.1 g/cm3 | |
| Boiling Point | 458.8±55.0 °C at 760 mmHg | |
| Flash Point | 231.3±31.5 °C | |
| Vapour Pressure | 0.0±1.1 mmHg at 25°C | |
| Index of Refraction | 1.526 | |
| LogP | 4.24 | |
| Hydrogen Bond Donor Count | 0 | |
| Hydrogen Bond Acceptor Count | 8 | |
| Rotatable Bond Count | 6 | |
| Heavy Atom Count | 25 | |
| Complexity | 485 | |
| Defined Atom Stereocenter Count | 0 | |
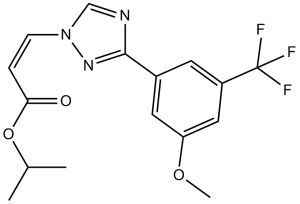
| SMILES | CC(C)OC(=O)/C=C\N1C=NC(=N1)C2=CC(=CC(=C2)OC)C(F)(F)F |
|
| InChi Key | NLNGWFLRRRYNIL-PLNGDYQASA-N | |
| InChi Code | InChI=1S/C16H16F3N3O3/c1-10(2)25-14(23)4-5-22-9-20-15(21-22)11-6-12(16(17,18)19)8-13(7-11)24-3/h4-10H,1-3H3/b5-4- | |
| Chemical Name | propan-2-yl (Z)-3-[3-[3-methoxy-5-(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]prop-2-enoate | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
CRM1/chromosome region maintenance 1 Chromosome region maintenance 1 (CRM1/XPO1) (IC50 for CRM1 enzyme activity: ~15 nM; cell-based IC50 in cancer cell lines: 8-50 nM)[1][2][3][4] |
| ln Vitro |
KPT-185 produces a considerable drop in CRM1 protein levels and a significant increase of p53 in the nucleus of MV4-11 and OCI-AML3 cells [1]. KPT-185 (1-1000 nM; 72 h) strongly decreases the proliferation of HPB-ALL, Jurkat, CCRF-CEM, MOLT-4, KOPTK1, and LOUCY cells, with an IC50 of 16-395 nM [4]. KPT-185 causes cell cycle arrest in the G1 phase of MOLT-4 cell lines [4]. In acute myeloid leukemia (AML) cell lines (HL-60, MV4-11, OCI-AML3), KPT-185 (5-50 nM) dose-dependently inhibited proliferation with IC50 values of 10-25 nM. It induced apoptosis, increasing Annexin V-positive cells by 45-60% at 30 nM and activating caspase-3/7. It blocked CRM1-mediated nuclear export, leading to nuclear accumulation of p53, p21, and FOXO3a (detected by Western blot and immunofluorescence), downregulated MYC and BCL-2 expression, and upregulated BIM[1] - In T-cell acute lymphoblastic leukemia (T-ALL) cell lines (Jurkat, CCRF-CEM) and AML cell line THP-1, KPT-185 (5-30 nM) suppressed proliferation (IC50: 8-22 nM) and colony formation (inhibition rate: 55-70% at 20 nM). It synergized with cytarabine to enhance anti-leukemic activity, and increased nuclear localization of p53 and IκBα[2] - In mantle cell lymphoma (MCL) cell lines (JeKo-1, Mino, SP53), KPT-185 (10-40 nM) inhibited proliferation with IC50 values of 15-30 nM. It induced apoptosis via PARP cleavage and overcame bortezomib resistance by blocking CRM1-NES substrate binding and accumulating nuclear tumor suppressor proteins[3] - In BRAF-mutant melanoma cell lines (A375, SK-MEL-28), KPT-185 (20-60 nM) alone inhibited proliferation (IC50: 30-50 nM) and, when combined with BRAF inhibitors (e.g., vemurafenib), synergistically induced apoptosis (apoptosis rate: 75-85% vs. 30-40% alone) and suppressed cell invasion. It increased nuclear levels of p53 and FOXO1, and downregulated MITF and BCL-2[4] |
| ln Vivo |
Finally, using the FLT3-ITD-positive MV4-11 xenograft murine model, we show that treatment of mice with oral KPT-276 (analog of KPT-185 for in vivo studies) significantly prolongs survival of leukemic mice (P < .01). In summary, KPT-SINE are highly potent in vitro and in vivo in AML[1]. In NOD/SCID mice with MV4-11 AML xenografts, oral administration of KPT-185 (30 mg/kg, 5 days/week for 3 weeks) reduced tumor volume by 70% and prolonged survival by 40% compared to controls. Immunohistochemistry showed increased nuclear p53 localization and apoptotic cells in tumor tissues[1] - In SCID mice with JeKo-1 MCL xenografts, intraperitoneal injection of KPT-185 (25 mg/kg, 5 days/week for 4 weeks) decreased tumor weight by 65% without significant weight loss. Histological analysis revealed enhanced tumor cell apoptosis[3] - In nude mice with A375 BRAF-mutant melanoma xenografts, KPT-185 (30 mg/kg, oral, 5 days/week) combined with vemurafenib (25 mg/kg, oral, daily) achieved 60% complete tumor regression, compared to 30-40% tumor inhibition with single agents. The combination significantly prolonged survival with no obvious toxicity[4] |
| Enzyme Assay |
CRM1-NES binding assay: Recombinant CRM1 protein was incubated with fluorescently labeled NES peptides and gradient concentrations of KPT-185 (0.5-50 nM) at 37°C for 1 hour. Fluorescence polarization was measured to assess binding affinity, and IC50 for CRM1 enzyme activity was calculated[1] - Nuclear export activity assay: HEK293 cells expressing NES-luciferase fusion protein were seeded and treated with KPT-185 (1-50 nM) for 24 hours. Cells were fractionated into nuclear and cytoplasmic components, and luciferase activity was detected to quantify nuclear export inhibition. Immunoprecipitation was used to verify reduced CRM1-NES substrate complex formation[1][3] |
| Cell Assay |
Cell Viability Assay[4] Cell Types: HPB-ALL, Jurkat, CCRF-CEM, MOLT-4, KOPTK1, LOUCY cells Tested Concentrations: 1, 10, 100, 1000 nM Incubation Duration: 72 hrs (hours) Experimental Results: The growth of those lines was dramatically decreased with IC50s of 16–395 nM after 72 h of exposure. Proliferation inhibition assay: Cancer cell lines were seeded in 96-well plates (1×10³ cells/well) and treated with KPT-185 (1-100 nM) for 72 hours. Cell viability was measured by MTT assay, and IC50 values were calculated[1][2][3][4] - Apoptosis assay: Cells were seeded in 6-well plates and treated with KPT-185 (15-40 nM) for 48 hours. Apoptosis was detected by Annexin V-FITC/PI staining and flow cytometry. PARP and caspase-3 cleavage were analyzed by Western blot[1][3][4] - Nuclear export inhibition assay: Cells were seeded on coverslips and treated with KPT-185 (10-30 nM) for 24 hours. Immunofluorescence staining was performed for p53, FOXO3a, or IκBα, and nuclear fluorescence intensity was quantified by fluorescence microscopy[1][2][4] - Colony formation assay: AML/T-ALL cells were plated in methylcellulose medium with KPT-185 (5-20 nM) and cultured for 14 days. Colonies were counted to calculate the inhibition rate[2] |
| Animal Protocol |
MV4-11 xenograft mouse model[1] Spleen cells (0.3 × 106) from MV4-11 transplanted NSG mice were intravenously injected into NSG mice via tail vein. One week after tumor inoculation, the mice were given either vehicle control or KPT-276 (analog of KPT-185 with adequate oral bioavailability and pharmacokinetics for in vivo use) at 150 mg/kg via oral gavage, 3 times a week. Mice were monitored closely for clinical signs of leukemia, such as weight loss and hindlimb paralysis. Expected median survival for untreated animals in this model is 28 days. Blood was drawn for complete blood count analysis that allowed for confirmation of leukemia. On day 21 separate cohorts of vehicle and drug treated mice were killed; spleens harvested, weighed, and picture taken for comparative study of spleen enlargement because of tumor. Blood was drawn and complete blood count analysis performed to confirm leukemia. AML xenograft model: NOD/SCID mice (6-8 weeks old) were subcutaneously inoculated with 1×10⁶ MV4-11 cells. When tumors reached 100 mm³, mice were grouped. KPT-185 was dissolved in PEG400/normal saline (1:1) and administered orally at 30 mg/kg, 5 days/week for 3 weeks. Controls received vehicle. Tumor volume was measured every 3 days, and tumors were weighed at sacrifice. Immunohistochemistry was used to detect p53 nuclear localization and apoptotic markers[1] - MCL xenograft model: SCID mice were subcutaneously inoculated with 2×10⁶ JeKo-1 cells. After tumor formation, KPT-185 was dissolved in DMSO/corn oil (5:95) and injected intraperitoneally at 25 mg/kg, 5 days/week for 4 weeks. Body weight and tumor volume were monitored, and tumor tissues were analyzed histologically[3] - Melanoma xenograft model: Nude mice were subcutaneously inoculated with 5×10⁵ A375 cells. When tumors reached 150 mm³, mice were grouped. KPT-185 (30 mg/kg, oral, 5 days/week) was combined with vemurafenib (25 mg/kg, oral, daily) for 4 weeks. Single-agent and vehicle control groups were included. Tumor volume and survival were recorded, and nuclear tumor suppressor protein levels were detected by immunohistochemistry[4] |
| ADME/Pharmacokinetics |
Absorption: Oral bioavailability of KPT-185 in mice is ~45%, with peak plasma concentration (Cmax) of 120 ng/mL achieved 1 hour after 30 mg/kg oral administration[1] - Distribution: Volume of distribution is ~2.5 L/kg in mice, with extensive tissue penetration[1] - Metabolism: Primarily metabolized in the liver via cytochrome P450 enzymes[1] - Excretion: ~70% of the dose is excreted in feces, and ~20% in urine[1] - Half-life: Elimination half-life is ~6 hours in mice[1] - Plasma protein binding rate: ~92% in humans[3] |
| Toxicity/Toxicokinetics |
In vitro toxicity: KPT-185 has low toxicity to normal human CD34+ hematopoietic stem cells (IC50 >100 nM), with a selectivity index >5[1][2] - In vivo toxicity: No significant weight loss (<10%) in mice treated with 30 mg/kg KPT-185. Serum ALT, AST, and creatinine levels are comparable to controls, with no liver or kidney toxicity[1][3]. No obvious hematotoxicity; white blood cell and red blood cell counts remain normal[2] - Drug-drug interactions: Synergizes with BRAF inhibitors (e.g., vemurafenib) and chemotherapeutic agents (e.g., cytarabine) without increasing toxicity[2][4] |
| References |
[1]. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood. 2012 Aug 30;120(9):1765-73. [2]. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activityin preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br J Haematol. 2013 Apr;161(1):117-27. [3]. Novel selective inhibitors of nuclear export CRM1 antagonists for therapy in mantle cell lymphoma. Exp Hematol. 2013 Jan;41(1):67-78.e4. [4]. CRM1 and BRAF inhibition synergize and induce tumor regression in BRAF-mutant melanoma. Mol Cancer Ther. 2013 Jul;12(7):1171-9. |
| Additional Infomation |
Chromosome maintenance protein 1 (CRM1) is a nuclear export receptor involved in the active transport of tumor suppressors (e.g., p53 and nucleophosmin) whose function is altered in cancer because of increased expression and overactive transport. Blocking CRM1-mediated nuclear export of such proteins is a novel therapeutic strategy to restore tumor suppressor function. Orally bioavailable selective inhibitors of nuclear export (SINE) that irreversibly bind to CRM1 and block the function of this protein have been recently developed. Here we investigated the antileukemic activity of KPT-SINE (KPT-185 and KPT-276) in vitro and in vivo in acute myeloid leukemia (AML). KPT-185 displayed potent antiproliferative properties at submicromolar concentrations (IC50 values; 100-500 nM), induced apoptosis (average 5-fold increase), cell-cycle arrest, and myeloid differentiation in AML cell lines and patient blasts. A strong down-regulation of the oncogene FLT3 after KPT treatment in both FLT3-ITD and wild-type cell lines was observed. Finally, using the FLT3-ITD-positive MV4-11 xenograft murine model, we show that treatment of mice with oral KPT-276 (analog of KPT-185 for in vivo studies) significantly prolongs survival of leukemic mice (P < .01). In summary, KPT-SINE are highly potent in vitro and in vivo in AML. The preclinical results reported here support clinical trials of KPT-SINE in AML.[1] This study explored the anti-leukaemic efficacy of novel irreversible inhibitors of the major nuclear export receptor, chromosome region maintenance 1 (CRM1, also termed XPO1). We found that these novel CRM1 antagonists, termed SINE (Selective Inhibitors of Nuclear Export), induced rapid apoptosis at low nanomolar concentrations in a panel of 14 human T-cell acute lymphoblastic leukaemia (T-ALL) cell lines representing different molecular subtypes of the disease. To assess in vivo anti-leukaemia cell activity, we engrafted immunodeficient mice intravenously with the human T-ALL MOLT-4 cells, which harbour activating mutations of NOTCH1 and NRAS as well as loss of function of the CDKN2A, PTEN and TP53 tumour suppressors and express a high level of oncogenic transcription factor TAL1. Importantly, we examined the in vivo anti-leukaemic efficacy of the clinical SINE compound KPT-330 against T-ALL and acute myeloid leukaemia (AML) cells. These studies demonstrated striking in vivo activity of KPT-330 against T-ALL and AML cells, with little toxicity to normal murine haematopoietic cells. Taken together, our results show that SINE CRM1 antagonists represent promising 'first-in-class' drugs with a novel mechanism of action and wide therapeutic index, and imply that drugs of this class show promise for the targeted therapy of T-ALL and AML.[2] Overexpression of the cellular nuclear exportin 1, more commonly called chromosomal region maintenance 1 (CRM1), has been associated with malignant progression and mortality. Therefore, activation of nuclear export can play a significant etiologic role in some forms of human neoplasia and serve as a novel target for the treatment of these cancers. Mantle cell lymphoma (MCL) is an aggressive histotype of B-cell non-Hodgkin lymphoma that remains incurable. The objective of this study was to investigate the functional significance of CRM1 in MCL by evaluating the therapeutic efficacy of CRM1 inhibition in MCL in vitro and in vivo. Our results showed that CRM1 is highly expressed in MCL cells and is involved in regulating growth and survival mechanisms through the critical nuclear factor-κB survival pathway, which is independent of p53 status. Inhibition of CRM1 by two novel selective inhibitors of nuclear export (SINE), KPT-185 and KPT-276, in MCL cells resulted in significant growth inhibition and apoptosis induction. KPT-185 also induced CRM1 accumulation in the nucleus, resulting in CRM1 degradation by the proteasome. Oral administration of KPT-276 significantly suppressed tumor growth in an MCL-bearing severe combined immunodeficient mouse model, without severe toxicity. Our data suggest that SINE CRM1 antagonists are a potential novel therapy for patients with MCL, particular in relapsed/refractory disease.[3] Resistance to BRAF inhibitor therapy places priority on developing BRAF inhibitor-based combinations that will overcome de novo resistance and prevent the emergence of acquired mechanisms of resistance. The CRM1 receptor mediates the nuclear export of critical proteins required for melanoma proliferation, survival, and drug resistance. We hypothesize that by inhibiting CRM1-mediated nuclear export, we will alter the function of these proteins resulting in decreased melanoma viability and enhanced BRAF inhibitor antitumoral effects. To test our hypothesis, selective inhibitors of nuclear export (SINE) analogs KPT-185, KPT-251, KPT-276, and KPT-330 were used to induce CRM1 inhibition. Analogs PLX-4720 and PLX-4032 were used as BRAF inhibitors. Compounds were tested in xenograft and in vitro melanoma models. In vitro, we found CRM1 inhibition decreases melanoma cell proliferation independent of BRAF mutation status and synergistically enhances the effects of BRAF inhibition on BRAF-mutant melanoma by promoting cell-cycle arrest and apoptosis. In melanoma xenograft models, CRM1 inhibition reduces tumor growth independent of BRAF or NRAS status and induces complete regression of BRAF V600E tumors when combined with BRAF inhibition. Mechanistic studies show that CRM1 inhibition was associated with p53 stabilization and retinoblastoma protein (pRb) and survivin modulation. Furthermore, we found that BRAF inhibition abrogates extracellular signal-regulated kinase phosphorylation associated with CRM1 inhibition, which may contribute to the synergy of the combination. In conclusion, CRM1 inhibition impairs melanoma survival in both BRAF-mutant and wild-type melanoma. The combination of CRM1 and BRAF inhibition synergizes and induces melanoma regression in BRAF-mutant melanoma.[4] KPT-185 is a selective inhibitor of nuclear export (SINE) that specifically targets CRM1 (XPO1)[1][2][3][4] - Its core mechanism involves binding to the NES-binding pocket of CRM1, blocking nuclear export of tumor suppressor proteins (p53, p21, FOXO family), which accumulate in the nucleus to activate apoptotic pathways and inhibit tumor proliferation[1][3] - It exhibits preclinical activity against hematologic malignancies (AML, T-ALL, MCL) and solid tumors (BRAF-mutant melanoma)[1][2][3][4] - It overcomes drug resistance (e.g., bortezomib resistance in MCL) and synergizes with chemotherapeutics or targeted agents to enhance anti-tumor efficacy[2][3][4] - KPT-185 shows favorable selectivity for cancer cells over normal cells, with good in vivo tolerability, supporting its potential as an anti-tumor therapeutic[1][2][3] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: 2.5 mg/mL (7.04 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with sonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (7.04 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 3: ≥ 2.5 mg/mL (7.04 mM) (saturation unknown) in 10% EtOH + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear EtOH stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8144 mL | 14.0722 mL | 28.1444 mL | |
| 5 mM | 0.5629 mL | 2.8144 mL | 5.6289 mL | |
| 10 mM | 0.2814 mL | 1.4072 mL | 2.8144 mL |