JNJ-38877605 (JNJ38877605) is an orally bioavailable small-molecule inhibitor of c-Met with potential antitumor activity. It exhibits 600-fold selectivity for inhibiting c-Met over 200 other kinases and inhibits it with an IC50 of 4 nM.
Physicochemical Properties
| Molecular Formula | C19H13F2N7 | |
| Molecular Weight | 377.35 | |
| Exact Mass | 377.12 | |
| Elemental Analysis | C, 60.48; H, 3.47; F, 10.07; N, 25.98 | |
| CAS # | 943540-75-8 | |
| Related CAS # |
|
|
| PubChem CID | 46911863 | |
| Appearance | Off-white to light yellow solid powder | |
| Density | 1.5±0.1 g/cm3 | |
| Index of Refraction | 1.734 | |
| LogP | 3.13 | |
| Hydrogen Bond Donor Count | 0 | |
| Hydrogen Bond Acceptor Count | 7 | |
| Rotatable Bond Count | 3 | |
| Heavy Atom Count | 28 | |
| Complexity | 564 | |
| Defined Atom Stereocenter Count | 0 | |
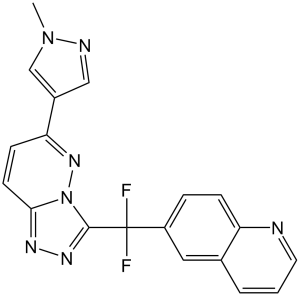
| SMILES | FC(C1N2C(C=CC(C3=CN(C)N=C3)=N2)=NN=1)(C1C=C2C(N=CC=C2)=CC=1)F |
|
| InChi Key | JRWCBEOAFGHNNU-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C19H13F2N7/c1-27-11-13(10-23-27)16-6-7-17-24-25-18(28(17)26-16)19(20,21)14-4-5-15-12(9-14)3-2-8-22-15/h2-11H,1H3 | |
| Chemical Name | 6-[difluoro-[6-(1-methylpyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-yl]methyl]quinoline | |
| Synonyms | JNJ38877605; JNJ 38877605; JNJ-38877605 | |
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | c-Met (IC50 = 4 nM) |
| ln Vitro | JNJ-38877605 potently inhibits HGF-stimulated and constitutively activated c-Met phosphorylation in vitro and exhibits more than 600-fold selectivity for c-Met when compared to more than 200 other diverse tyrosine and serine-threonine kinases.[1] One important factor in invasive growth that is significantly reduced in EBC1, GTL16, NCI-H1993, and MKN45 cells is JNJ-38877605 (500 nM) phosphorylation of Met and RON.[2] uPAR, IL-6, GROa, and IL-8 secretion in GTL16 cells are all modulated by JNJ-38877605, according to a recent study. |
| ln Vivo | JNJ-38877605, when administered orally at 40 mg/kg/day for 72 hours in mice with established GTL16 xenografts, causes a statistically significant reduction in human IL-8 plasma levels (from 0.150 ng/mL to 0.050 ng/mL) and GROα plasma levels (from 0.080 ng/mL to 0.030 ng/mL). At the same dosage, however, blood levels of uPAR drop to levels greater than 50%.[3] |
| Enzyme Assay | JNJ-38877605 is an ATP-competitive inhibitor of c-Met with an IC50 of 4 nM that is 600 times more selective for c-Met than 200 other tyrosine and serine-threonine substrates. |
| Animal Protocol |
GTL16 cells are inoculated subcutaneously into the right posterior flank (or both right and left posterior flanks, for determination of uPAR and IL-6) of 6-week-old immunodeficient nu/nu female mice on Swiss CD1 background. ≤40 mg/kg/day Administered via p.o. |
| References |
[1]. JNJ-38877605: a selective Met kinase inhibitor inducing regression of Met-driven tumor models. [2]. J Natl Cancer Inst . 2011 Apr 20;103(8):645-61. [3]. Int J Cancer . 2012 Mar 15;130(6):1357-66. |
| Additional Infomation |
6-[difluoro-[6-(1-methyl-4-pyrazolyl)-[1,2,4]triazolo[4,3-b]pyridazin-3-yl]methyl]quinoline is a member of quinolines. JNJ-38877605 has been used in trials studying the treatment of Neoplasms. c-Met Inhibitor JNJ-38877605 is an orally bioavailable, small-molecule receptor tyrosine kinase inhibitor with potential antineoplastic activity. c-Met inhibitor JNJ-38877605 selectively inhibits c-Met, a receptor tyrosine kinase (RTK) involved in cancer cell survival and invasiveness, and tumor angiogenesis. c-Met is also known as hepatocyte growth factor receptor (HGFR). |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (5.51 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (5.51 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (5.51 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 30% propylene glycol, 5% Tween 80, 65% D5W: 30 mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6501 mL | 13.2503 mL | 26.5006 mL | |
| 5 mM | 0.5300 mL | 2.6501 mL | 5.3001 mL | |
| 10 mM | 0.2650 mL | 1.3250 mL | 2.6501 mL |