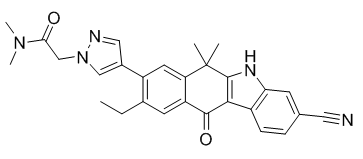
JH-VIII-157-02, an alectinib analogue, is a novel and potent ALK inhibitor with anticancer activities. It inhibits ALK with an IC50 of 2 nM for echinoderm microtubule-associated protein-like 4-ALK (EML4-ALK) G1202R in cell assays.
Physicochemical Properties
| Molecular Formula | C28H27N5O2 |
| Molecular Weight | 465.546285867691 |
| Exact Mass | 465.216 |
| CAS # | 1639422-97-1 |
| PubChem CID | 127037084 |
| Appearance | White to off-white solid powder |
| LogP | 4.5 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 4 |
| Rotatable Bond Count | 4 |
| Heavy Atom Count | 35 |
| Complexity | 893 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | O=C1C2C=C(CC)C(C3C=NN(CC(N(C)C)=O)C=3)=CC=2C(C)(C)C2=C1C1C=CC(C#N)=CC=1N2 |
| InChi Key | IDGNCVHQDDOPND-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C28H27N5O2/c1-6-17-10-21-22(11-20(17)18-13-30-33(14-18)15-24(34)32(4)5)28(2,3)27-25(26(21)35)19-8-7-16(12-29)9-23(19)31-27/h7-11,13-14,31H,6,15H2,1-5H3 |
| Chemical Name | 2-[4-(3-cyano-9-ethyl-6,6-dimethyl-11-oxo-5H-benzo[b]carbazol-8-yl)pyrazol-1-yl]-N,N-dimethylacetamide |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Structural analogue JH-VIII-157-02 functions as an ALK inhibitor against echinoderm microtubule-associated protein-like 4-ALK (EML4-ALK) G1202R in cells, with an IC50 of 2 nM. Additionally, EML4-ALKwt (Eawt), EAC1156Y, EAF1174L, EAS1206Y (IC50, 2 nM), EAG1269A (IC50, 3 nM), EAL1196M (IC50, 58 nM), EA1151Tins (IC50, 107 nM), and EAL1152R (IC50, 196 nM) are all effectively inhibited by JH-VIII-157-02. JH-VIII-157-02 is also selective for other kinases, with IC50 values of 14 nM, 14 nM, 3 nM, 13 nM, and 12 nM for IRAK1, CLK4, RET, RET V804L, RET V804M, and IRAK 4. Both 465nM and 465nM are. H3122 and DFCI76 (L1152R), two cancer cell lines, are inhibited by JH-VIII-157-02, with EC50s of 5 and 19 nM, respectively [1]. |
| ln Vitro |
Structural analogue JH-VIII-157-02 functions as an ALK inhibitor against echinoderm microtubule-associated protein-like 4-ALK (EML4-ALK) G1202R in cells, with an IC50 of 2 nM. Additionally, EML4-ALKwt (Eawt), EAC1156Y, EAF1174L, EAS1206Y (IC50, 2 nM), EAG1269A (IC50, 3 nM), EAL1196M (IC50, 58 nM), EA1151Tins (IC50, 107 nM), and EAL1152R (IC50, 196 nM) are all effectively inhibited by JH-VIII-157-02. JH-VIII-157-02 is also selective for other kinases, with IC50 values of 14 nM, 14 nM, 3 nM, 13 nM, and 12 nM for IRAK1, CLK4, RET, RET V804L, RET V804M, and IRAK 4. Both 465nM and 465nM are. H3122 and DFCI76 (L1152R), two cancer cell lines, are inhibited by JH-VIII-157-02, with EC50s of 5 and 19 nM, respectively [1]. JH-VIII-157-02 (Compound 6) potently inhibited the proliferation of Ba/F3 cells transformed with EML4-ALK wild type and various crizotinib-resistant secondary mutants (C1156Y, F1174L, L1196M, L1152R, 1151Tins, G1202R, G1269A, S1206Y), with IC50 values ranging from 2 to 196 nM. It was particularly potent against the G1202R mutant (IC50 = 2 nM), representing a 100-fold increase in cellular potency compared to alectinib. [1] In NIH-3T3 cells transformed with wild-type EML4-ALK or its crizotinib-resistant mutants, JH-VIII-157-02 strongly inhibited phospho-ALK levels as demonstrated by western blotting. [1] The compound showed submicromolar antiproliferative EC50 values across a panel of NSCLC cell lines (H3122: 5 nM; DFC176 (L1152R): 19 nM; DFC114 (G1269A): 419 nM) and neuroblastoma cell lines (e.g., Kelly (F1174L): 147 nM; LAN-5 (R1275Q): 192 nM). [1] Kinome-wide selectivity profiling at 1 μM revealed JH-VIII-157-02 interacted with 34 kinases, indicating moderate selectivity with an S(1) score of 0.06. Significant off-target activities included potent inhibition of RET, IRAK1, IRAK4, and CLK4. [1] |
| ln Vivo | At an oral dose of 10 mg/kg, JH-VIII-157-02 showed good oral bioavailability in mice. JH-VIII-157-02 is also capable of penetrating mice's central nervous systems [1]. |
| Cell Assay |
Cellular Viability/Proliferation Assay (MTS): Ba/F3 cells transformed with EML4-ALK (wild type or mutants) or untransduced Ba/F3 control cells were treated with JH-VIII-157-02 in a dose-escalation manner. After 72 hours, cell viability was assessed using the MTS assay. IC50 values were calculated by nonlinear regression. [1] Cellular Viability/Proliferation Assay (CellTiter-Glo): NSCLC and neuroblastoma cell lines were seeded in 96-well plates and exposed to JH-VIII-157-02 in triplicate at concentrations ranging from 1 nM to 10 μM for 72 hours. Cell viability was evaluated using the CellTiter-Glo Luminescent Cell Viability Assay. EC50 values were calculated by nonlinear regression. [1] Western Blotting: NIH-3T3 cells transformed with wild-type EML4-ALK or its crizotinib-resistant mutants were treated with JH-VIII-157-02. Cells were lysed, and proteins were separated by SDS-PAGE, transferred to a membrane, and probed with an anti-phospho-ALK antibody to assess target inhibition. [1] KINOMEscan Selectivity Profiling: JH-VIII-157-02 was screened at a concentration of 1 μM against a panel of 456 kinases using the KINOMEscan competition-binding assay. Dose-response analyses were performed for identified off-target kinases to determine IC50 values. [1] |
| Animal Protocol |
Pharmacokinetic Study: Mice were administered JH-VIII-157-02 intravenously (2 mg/kg) or orally (10 mg/kg). Blood samples were collected at various time points. Plasma was separated, and compound concentrations were determined using LC-MS/MS. Pharmacokinetic parameters (half-life, Cmax, AUC, bioavailability) were calculated. [1] Central Nervous System (CNS) Penetration Study: Mice were administered JH-VIII-157-02 intravenously (2 mg/kg) or orally (10 mg/kg). At 2 and 8 hours post-dose, animals were euthanized. Plasma and brain tissues were collected, homogenized, and analyzed for compound concentration using LC-MS/MS. Brain-to-plasma concentration ratios were calculated. [1] |
| ADME/Pharmacokinetics |
Following a single intravenous dose of 2 mg/kg in mice, JH-VIII-157-02 had a plasma half-life (T1/2) of 1.64 hours, a maximum plasma concentration (Cmax) of 75.71 μM, and an area under the curve (AUC last) of 27.56 μMhr. [1] Following a single oral dose of 10 mg/kg in mice, JH-VIII-157-02 had a plasma half-life (T1/2) of 1.69 hours, a Cmax of 0.80 μM, a Tmax of 0.83 hours, an AUC last of 2.31 μMhr, and an oral bioavailability (F) of 87%. [1] Two hours after a 10 mg/kg oral dose, the plasma concentration was 0.34 μM, and the brain concentration was 0.03 μM, resulting in a total brain-to-plasma concentration ratio of 0.1. [1] |
| Toxicity/Toxicokinetics |
In untransduced Ba/F3 cells (cytotoxicity control), JH-VIII-157-02 showed an IC50 of 591 nM, indicating a selectivity window over ALK-driven cells. [1] |
| References |
[1]. Discovery of Inhibitors That Overcome the G1202R Anaplastic Lymphoma Kinase Resistance Mutation. J Med Chem. 2015 Dec 10;58(23):9296-9308. |
| Additional Infomation |
JH-VIII-157-02 is a structural analogue of the ALK inhibitor alectinib, designed to overcome the G1202R resistance mutation. Molecular modeling suggested it forms key hydrogen bond interactions with the guanidine moiety of R1202 in the mutant and with R1120 in wild-type ALK. [1] It demonstrated good oral bioavailability and moderate CNS penetration in mice. [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ≥ 25 mg/mL (~53.70 mM) |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1480 mL | 10.7400 mL | 21.4800 mL | |
| 5 mM | 0.4296 mL | 2.1480 mL | 4.2960 mL | |
| 10 mM | 0.2148 mL | 1.0740 mL | 2.1480 mL |