Physicochemical Properties
| Molecular Formula | C18H24CLN3O2 |
| Molecular Weight | 349.855063438416 |
| Exact Mass | 349.156 |
| Elemental Analysis | C, 61.80; H, 6.91; Cl, 10.13; N, 12.01; O, 9.15 |
| CAS # | 1005398-61-7 |
| Related CAS # | Irdabisant;1005402-19-6 |
| PubChem CID | 53363810 |
| Appearance | Typically exists as solid at room temperature |
| LogP | 3.842 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 4 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 24 |
| Complexity | 467 |
| Defined Atom Stereocenter Count | 1 |
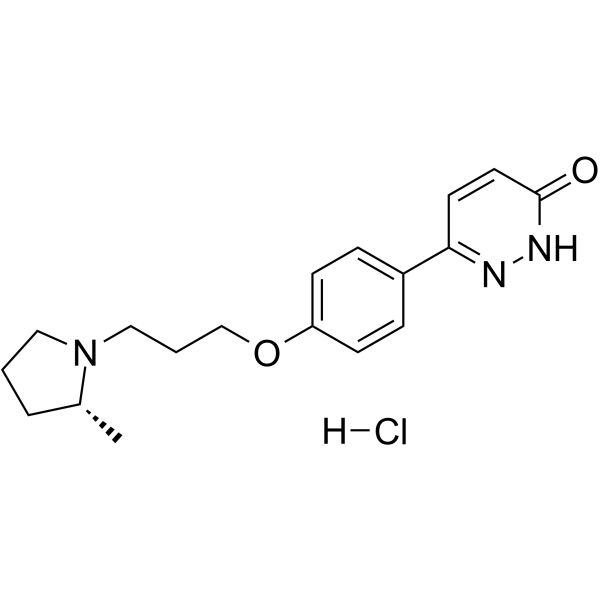
| SMILES | Cl.O(C1C=CC(C2C=CC(NN=2)=O)=CC=1)CCCN1CCC[C@H]1C |
| InChi Key | WJUJICMNSMPLLG-PFEQFJNWSA-N |
| InChi Code | InChI=1S/C18H23N3O2.ClH/c1-14-4-2-11-21(14)12-3-13-23-16-7-5-15(6-8-16)17-9-10-18(22)20-19-17;/h5-10,14H,2-4,11-13H2,1H3,(H,20,22);1H/t14-;/m1./s1 |
| Chemical Name | 3-[4-[3-[(2R)-2-methylpyrrolidin-1-yl]propoxy]phenyl]-1H-pyridazin-6-one;hydrochloride |
| Synonyms | IRDABISANT HYDROCHLORIDE; 1005398-61-7; Irdabisant hydrochloride [USAN]; CEP-26401 HYDROCHLORIDE; UNII-M2U80Z023U; M2U80Z023U; Irdabisant hydrochloride (USAN); 3(2H)-Pyridazinone, 6-(4-(3-((2R)-2-methyl-1-pyrrolidinyl)propoxy)phenyl)-, hydrochloride (1:1); |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
- Histamine H₃ receptor (H₃R) (Ki = 0.54 nM in human recombinant H₃R radioligand binding assay; IC₅₀ = 2.3 nM in rat brain H₃R binding assay) [1] - Histamine H₃ receptor (H₃R) (selective for H₃R over other histamine receptors: Ki for H₁R > 10,000 nM, H₂R > 10,000 nM, H₄R > 10,000 nM; no significant binding to 50+ other GPCRs, ion channels, or enzymes at 10 μM) [2] |
| ln Vitro |
1. H₃R Inverse Agonist Activity:
In human recombinant H₃R-expressing CHO cells, CEP-26401 (irdabisant) inhibited basal [³⁵S]-GTPγS binding (a measure of H₃R constitutive activity) with IC₅₀ = 1.8 nM; it also antagonized histamine-induced [³⁵S]-GTPγS binding with Ki = 0.62 nM. This confirmed inverse agonist activity at H₃R [1] 2. Neurotransmitter Release Promotion: In rat cortical synaptosomes, CEP-26401 (irdabisant) (10 nM–1 μM) dose-dependently increased K⁺-evoked release of acetylcholine (maximal 2.1-fold increase at 100 nM) and histamine (maximal 1.8-fold increase at 100 nM). The effect was blocked by the selective H₃R antagonist ciproxifan, confirming H₃R-mediated action [2] 3. Receptor Selectivity: Screening against 56 unrelated receptors (e.g., dopamine D₂, serotonin 5-HT₆), ion channels (e.g., Na⁺ channels), and enzymes (e.g., acetylcholinesterase) showed <5% binding inhibition by CEP-26401 (irdabisant) at 10 μM, demonstrating high H₃R selectivity [1] Compound 8a, Irdabisant (CEP-26401), exhibits inverse agonist activity with EC50 values of 2.0 nM and 1.1 nM against rat H3R and human H3R, respectively, and has Kb antagonist activity against both H3Rs with app values of 1.0 nM and 0.4 nM, respectively[1]. In addition to muscarinic M2 (Ki = 3.7 ± 0.0 μM) and adrenergic α1A (Ki = 9.8 ± 0.3 μM) receptors, irdabisant also exhibits activity against norepinephrine (Ki = 10 ± 1 μM), dopamine transporter (Ki = 11 ± 2 μM), and phosphodiesterase PDE3 (IC50 = 15 ± 1 μM) [1]. With IC50 values more than 30 μM, irdabisant inhibits the cytochrome P450 enzymes CYP1A2, 2C9, 2C19, 2D6, and 3A4 [1]. This suggests a low risk of drug interactions. |
| ln Vivo |
1. H₃R Occupancy in Rat Brain:
Oral administration of CEP-26401 (irdabisant) to rats at 0.1 mg/kg, 0.3 mg/kg, and 1 mg/kg resulted in 45%, 72%, and 91% H₃R occupancy in the cerebral cortex (measured via ex vivo [³H]-GSK189254 binding assay) at 2 hours post-dose. Occupancy persisted for 8 hours (32% at 8 hours with 1 mg/kg dose) [1] 2. Cognition-Enhancing Activity: In male Sprague-Dawley rats trained in the Morris water maze (spatial memory task), oral CEP-26401 (irdabisant) (0.3 mg/kg, 1 mg/kg) significantly reduced escape latency to the hidden platform by 38% and 52%, respectively, compared to vehicle. It also increased time spent in the target quadrant during probe trials (2.4-fold vs. vehicle at 1 mg/kg), indicating improved spatial memory [2] 3. Wake-Promoting Activity: In C57BL/6 mice instrumented for EEG/EMG recording, oral CEP-26401 (irdabisant) (1 mg/kg, 3 mg/kg) increased wake time by 18% and 31% over 6 hours post-dose, respectively, without altering sleep architecture (NREM/REM sleep ratio remained unchanged). The effect was absent in H₃R knockout mice, confirming H₃R dependency [2] The H3R agonist RAMH-induced smoking is dose-dependently inhibited by CEP-26401 (0.01-0.3 mg/kg; oral; single dose) [1]. In a social recognition short-term memory model, CEP-26401 (0.0001-0.1 mg/kg; intravenously or orally; single dose) enhances rat performance [1]. Rats treated orally with CEP-26401 (3–30 mg/kg; single dose) show wake-promoting effects [2]. In DBA/2NCrl mice, CEP-26401 (3–30 mg/kg; intraperitoneally) raises prepulse inhibition (PPI) [2]. CEP-26401 (1 mg/kg IV, 3 mg/kg PO; single dose) exhibits a moderate clearance rate in dogs and monkeys relative to rats rate, high oral bioavailability, and rapid absorption in rats and monkeys[1]. Irdabisant (compound 8a) pharmacokinetic parameters in rats, dogs, and monkeys [1]. Rat dog monkey IV t1/2 (h) 2.6 2.9 5.4 iv Vd (L/kg) 9.4 3.5 ± 1.1 3.8 ± 0.9 iv CL (mL/min/kg) 42 13.2 ± 1.5 7.7 ± 1.8 po t1/2 (L /kg) 2.9 2.7 5.0 po AUC (ng·h/mL) 984 1190 ± 180 1919 ± 611 po Cmax (ng/mL) 270 230 ± 70 760 ± 74 po F (%) 83 22 ± 2 83 ± 18 Brain to Plasma ratio 2.6 ± 0.2 2.4 ± 0.4 / |
| Enzyme Assay |
1. Human Recombinant H₃R Radioligand Binding Assay:
Membranes from CHO cells expressing human H₃R were incubated with [³H]-GSK189254 (0.2 nM, a selective H₃R ligand) and serial dilutions of CEP-26401 (irdabisant) (0.01 nM–10 μM) in 50 mM Tris-HCl buffer (pH 7.4, containing 10 mM MgCl₂ and 0.1% BSA) at 25°C for 90 minutes. Non-specific binding was defined with 10 μM tiotidine (a non-selective H₃R antagonist). Bound radioactivity was separated by vacuum filtration through GF/B filters pre-soaked in 0.5% PEI, and counted via liquid scintillation. Ki values were calculated using the Cheng-Prusoff equation [1] 2. [³⁵S]-GTPγS Binding Assay (Inverse Agonist Activity): Membranes from human H₃R-CHO cells were incubated with CEP-26401 (irdabisant) (0.01 nM–10 μM), 0.1 nM [³⁵S]-GTPγS, and 10 μM GDP in 50 mM Tris-HCl buffer (pH 7.4, 10 mM MgCl₂, 100 mM NaCl) at 30°C for 60 minutes. For antagonist mode, 100 nM histamine (a H₃R agonist) was added to induce [³⁵S]-GTPγS binding. Bound radioactivity was filtered through GF/C filters and counted. IC₅₀ values were derived from sigmoidal dose-response curves [1] |
| Cell Assay |
1. Rat Cortical Synaptosome Neurotransmitter Release Assay:
Rat cerebral cortex was homogenized and synaptosomes were isolated via differential centrifugation. Synaptosomes were resuspended in Krebs-Ringer buffer (pH 7.4) and pre-incubated with CEP-26401 (irdabisant) (10 nM–1 μM) for 15 minutes. K⁺ (30 mM) was added to evoke neurotransmitter release, and samples were incubated for 5 minutes. Acetylcholine release was measured via choline acetyltransferase-coupled luminescence assay; histamine release was measured via ELISA. Vehicle-treated synaptosomes served as negative controls, and ciproxifan (1 μM) was used as a positive control for H₃R blockade [2] 2. CHO Cell H₃R Expression Validation: CHO cells stably expressing human H₃R were cultured in DMEM with 10% FBS. Cells were harvested, and H₃R expression was confirmed via flow cytometry using an anti-H₃R antibody. For functional assays, cells were seeded in 96-well plates (5×10⁴ cells/well) and serum-starved for 24 hours before treatment with CEP-26401 (irdabisant) [1] |
| Animal Protocol |
Animal/Disease Models: Male SD (SD (Sprague-Dawley)) rat (addiction model induced by intraperitoneal (ip) injection of 10 mg/kg RAMH) [1] Doses: 0.01-0.3 mg/kg Route of Administration: oral; single Dose Experimental Results: Dose-dependent inhibition of water drinking induced by the H3R agonist RAMH (shown as water drinking), with an EC50 value of 0.06 mg/kg. Animal/Disease Models: Male SD (SD (Sprague-Dawley)) rats (adult rats are briefly exposed to juvenile rats to establish a social recognition model) [2] Doses: intraperitoneal (ip) injection 0.0001, 0.001, 0.01 and 0.1 mg/kg; oral 0.01 and 0.1 mg/kg Route of Administration: iv or po; single-dose Experimental Results: effective rate reduction over study duration (RID) across the 0.001 to 0.1 mg/kg ip dose range and 0.01 to 0.1 mg/kg po dose range, demonstrating this model Effective enhancement of short- and medium-term sensory memory. Animal/Disease Models: Male SD (SD (Sprague-Dawley)) rat [2] Doses: 3, 10 and 30 mg/kg Route of Administration: Oral; Single Dose Experimental Results: 90% of treated animals within 3 hrs (hrs (hours)) of 30 mg/kg dose of time in the a 1. Rat Brain H₃R Occupancy Study: Male Sprague-Dawley rats (250–300 g) were fasted overnight and orally administered CEP-26401 (irdabisant) (0.1 mg/kg, 0.3 mg/kg, 1 mg/kg) dissolved in 0.5% methylcellulose/0.1% Tween 80. Vehicle-treated rats served as controls. At 2 hours or 8 hours post-dose, rats were euthanized, and cerebral cortex was dissected. Cortical membranes were prepared and incubated with [³H]-GSK189254 to measure unoccupied H₃R; occupancy was calculated as (1 – (bound radioactivity in treated rats / bound radioactivity in vehicle rats)) × 100% [1] 2. Rat Morris Water Maze (Spatial Memory) Study: Male Sprague-Dawley rats (300–350 g) were trained in a Morris water maze (1.8 m diameter, 60 cm depth) with a hidden platform (10 cm diameter) submerged 1 cm below water. Training sessions (4 trials/day, 60-second maximum trial time) were conducted for 5 days. On day 6, rats received oral CEP-26401 (irdabisant) (0.3 mg/kg, 1 mg/kg) or vehicle 1 hour before a probe trial (platform removed). Escape latency (time to find platform during training) and time spent in the target quadrant (during probe trial) were recorded via video tracking [2] 3. Mouse EEG/EMG Sleep-Wake Study: Male C57BL/6 mice (20–25 g) were implanted with EEG electrodes (frontal-parietal) and EMG electrodes (neck muscles) under anesthesia. After 7 days of recovery, mice were orally administered CEP-26401 (irdabisant) (1 mg/kg, 3 mg/kg) or vehicle at dark onset. EEG/EMG signals were recorded continuously for 6 hours, and sleep-wake states (wake, NREM sleep, REM sleep) were scored in 10-second epochs using sleep analysis software. H₃R knockout mice were used as a negative control group [2] |
| ADME/Pharmacokinetics |
- Oral Bioavailability: In rats, oral administration of CEP-26401 (irdabisant) (1 mg/kg) resulted in oral bioavailability of 68%, with Cₘₐₓ = 123 ng/mL and Tₘₐₓ = 1.5 hours. In beagle dogs, oral bioavailability was 75%, with Cₘₐₓ = 98 ng/mL and Tₘₐₓ = 2 hours [1] - Plasma Half-Life (t₁/₂): In rats, intravenous CEP-26401 (irdabisant) (0.3 mg/kg) had a plasma t₁/₂ of 3.2 hours; oral administration (1 mg/kg) had a t₁/₂ of 4.1 hours. In dogs, intravenous (0.1 mg/kg) and oral (0.3 mg/kg) t₁/₂ were 5.8 hours and 6.5 hours, respectively [1] - Plasma Protein Binding: CEP-26401 (irdabisant) showed 92% plasma protein binding in human plasma, 90% in rat plasma, and 88% in dog plasma (measured via equilibrium dialysis at 1 μM) [1] - Tissue Distribution: In rats, 1 hour after oral administration of CEP-26401 (irdabisant) (1 mg/kg), the brain-to-plasma concentration ratio was 0.8, indicating good blood-brain barrier penetration. Highest tissue concentrations were observed in liver (1200 ng/g) and kidney (850 ng/g), with low concentrations in muscle (120 ng/g) [2] - Metabolism: In human liver microsomes, CEP-26401 (irdabisant) was primarily metabolized via CYP3A4 (accounting for 70% of metabolism) and CYP2D6 (20%). No major active metabolites were detected; the main metabolite was a hydroxylated derivative with <10% H₃R affinity [1] |
| Toxicity/Toxicokinetics |
- Acute Toxicity: Oral administration of CEP-26401 (irdabisant) to rats at doses up to 2000 mg/kg showed no mortality or overt toxicity (e.g., no weight loss, no abnormal behavior) over 14 days. The LD₅₀ was determined to be >2000 mg/kg (oral) in rats and mice [1] - Subacute Toxicity: Rats were orally administered CEP-26401 (irdabisant) (1 mg/kg, 10 mg/kg, 100 mg/kg) daily for 28 days. No significant changes in body weight, food intake, or hematological parameters (e.g., RBC, WBC, platelets) were observed. Serum ALT and AST levels (markers of liver injury) were within normal ranges in all dose groups. Histopathological examination of liver, kidney, brain, and heart showed no treatment-related lesions [1] - Drug-Drug Interaction Potential: CEP-26401 (irdabisant) did not inhibit CYP1A2, CYP2C9, CYP2C19, CYP2D6, or CYP3A4 at concentrations up to 10 μM (in human liver microsomes), indicating low potential for interacting with drugs metabolized by these enzymes [1] |
| References |
[1]. Discovery and characterization of 6-{4-[3-(R)-2-methylpyrrolidin-1-yl)propoxy]phenyl}-2H-pyridazin-3-one (CEP-26401, irdabisant): a potent, selective histamine H3 receptor inverse agonist. J Med Chem. 2011 Jul 14;54(13):4781-92. [2]. CEP-26401 (irdabisant), a potent and selective histamine H₃ receptor antagonist/inverse agonist with cognition-enhancing and wake-promoting activities. J Pharmacol Exp Ther. 2012 Jan;340(1):124-33. |
| Additional Infomation |
- Background:
CEP-26401 (irdabisant) is a pyridazinone-derived H₃R inverse agonist developed to target cognitive deficits (e.g., in Alzheimer’s disease) and excessive sleepiness (e.g., in narcolepsy). H₃R is a presynaptic autoreceptor that inhibits histamine and other neurotransmitter release; inverse agonists reverse this inhibition to enhance neurotransmission [1] - Mechanism of Action: As an H₃R inverse agonist, CEP-26401 (irdabisant) binds to H₃R and reduces its constitutive activity, thereby increasing the release of histamine (from tuberomammillary nucleus) and other cognition-relevant neurotransmitters (acetylcholine, dopamine) in the brain. This enhances neuronal activity in brain regions involved in memory (e.g., hippocampus) and wakefulness (e.g., hypothalamus) [2] - Preclinical Efficacy Scope: Beyond spatial memory, CEP-26401 (irdabisant) (1 mg/kg, oral) improved working memory in rats (radial arm maze task, 40% reduction in error rate) and object recognition memory (35% increase in discrimination index). In a mouse model of narcolepsy, it reduced cataplexy episodes by 50% at 3 mg/kg (oral) [2] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8583 mL | 14.2914 mL | 28.5829 mL | |
| 5 mM | 0.5717 mL | 2.8583 mL | 5.7166 mL | |
| 10 mM | 0.2858 mL | 1.4291 mL | 2.8583 mL |