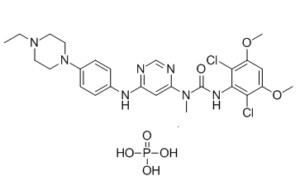
Infigratinib phosphate (formerly BGJ-398; BGJ398; NVP-BGJ398 phosphate; Truseltiq), the phosphate salt of Infigratinib, is an orally bioavailable FGFR (fibroblast growth factor receptors) inhibitor that has gained FDA approval in May 2021 to treat cholangiocarcinoma whose disease meets certain criteria.In cell-free experiments, it inhibits FGFR1/2/3 with IC50s of 0.9 nM/1.4 nM/1 nM, suggesting that it may have antiangiogenic and antineoplastic properties.
Physicochemical Properties
| Molecular Formula | C26H34CL2N7O7P | |
| Molecular Weight | 658.47 | |
| Exact Mass | 657.163 | |
| Elemental Analysis | C, 47.43; H, 5.20; Cl, 10.77; N, 14.89; O, 17.01; P, 4.70 | |
| CAS # | 1310746-10-1 | |
| Related CAS # | Infigratinib;872511-34-7 | |
| PubChem CID | 56669626 | |
| Appearance | White to off-white solid powder | |
| LogP | 4.574 | |
| Hydrogen Bond Donor Count | 5 | |
| Hydrogen Bond Acceptor Count | 12 | |
| Rotatable Bond Count | 8 | |
| Heavy Atom Count | 43 | |
| Complexity | 773 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | ClC1C(=C([H])C(=C(C=1N([H])C(N(C([H])([H])[H])C1C([H])=C(N=C([H])N=1)N([H])C1C([H])=C([H])C(=C([H])C=1[H])N1C([H])([H])C([H])([H])N(C([H])([H])C([H])([H])[H])C([H])([H])C1([H])[H])=O)Cl)OC([H])([H])[H])OC([H])([H])[H].P(=O)(O[H])(O[H])O[H] |
|
| InChi Key | GUQNHCGYHLSITB-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C26H31Cl2N7O3.H3O4P/c1-5-34-10-12-35(13-11-34)18-8-6-17(7-9-18)31-21-15-22(30-16-29-21)33(2)26(36)32-25-23(27)19(37-3)14-20(38-4)24(25)28;1-5(2,3)4/h6-9,14-16H,5,10-13H2,1-4H3,(H,32,36)(H,29,30,31);(H3,1,2,3,4) | |
| Chemical Name | 3-(2,6-dichloro-3,5-dimethoxyphenyl)-1-[6-[4-(4-ethylpiperazin-1-yl)anilino]pyrimidin-4-yl]-1-methylurea;phosphoric acid | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
FGFR1 (IC50 = 0.9 nM); FGFR2 (IC50 = 1.4 nM); FGFR3 (IC50 = 1 nM); FGFR4 (IC50 = 60 nM) Infigratinib phosphate suppresses FGFR1, FGFR2, and FGFR3 at IC50 values of ~1 nM, FGFR3K650E at IC50 values of 4.9 nM, and FGFR4 at IC50 values of 60 nM. With the exception of VEGFR2, KIT, and LYN, which are inhibited at submicromolar concentrations (IC50=0.18, 0.75, and 0.3 μM, respectively), the IC50 values for all other kinases fall within the μM range (FYN, LCK, YES, and ABL, IC50=1.9, 2.5, 1.1, and 2.3 μM, respectively). With IC50 values in the low nanomolar range, infigratinib inhibits the proliferation of the FGFR1-, FGFR2-, and FGFR3-dependent BaF3 cells in a manner similar to that seen in the enzymatic assay for the inhibition of receptor kinase activity. Except for VEGFR2 (IC50 1449 and 938 nM), which has at least a 400-fold selectivity versus FGFR1, FGFR2, and FGFR3, all IC50 values for the remaining cells are greater than 1.5 μM[1]. The growth of FGFR2-mutant endometrial cancer cells is effectively inhibited by infigratinib (at concentrations between 1 nM and 10 μM)[2]. |
| ln Vitro |
Infigratinib phosphate suppresses FGFR1, FGFR2, and FGFR3 at IC50 values of ~1 nM, FGFR3K650E at IC50 values of 4.9 nM, and FGFR4 at IC50 values of 60 nM. With the exception of VEGFR2, KIT, and LYN, which are inhibited at submicromolar concentrations (IC50=0.18, 0.75, and 0.3 μM, respectively), the IC50 values for all other kinases fall within the μM range (FYN, LCK, YES, and ABL, IC50=1.9, 2.5, 1.1, and 2.3 μM, respectively). With IC50 values in the low nanomolar range, infigratinib inhibits the proliferation of the FGFR1-, FGFR2-, and FGFR3-dependent BaF3 cells in a manner similar to that seen in the enzymatic assay for the inhibition of receptor kinase activity. Except for VEGFR2 (IC50 1449 and 938 nM), which has at least a 400-fold selectivity versus FGFR1, FGFR2, and FGFR3, all IC50 values for the remaining cells are greater than 1.5 μM[1]. The growth of FGFR2-mutant endometrial cancer cells is effectively inhibited by infigratinib (at concentrations between 1 nM and 10 μM)[2]. The selective FGFR inhibitor NVP-BGJ398 inhibited proliferation of all 20 tested endometrial cancer cell lines in a concentration-dependent manner, with IC50 values ranging from 1.0 µmol/L in the FGFR2-mutated AN3CA cell line to 10.0 µmol/L in the KRAS-mutated HEC1A cell line. Cell lines with activating FGFR2 mutations (S252W, N550K) had significantly lower mean IC50 values compared to FGFR2 wild-type cell lines (mean IC50 2.96 vs. 5.55, P = 0.021). The BRAF-mutated MFE319 cell line (which also harbors an FGFR2 S252W mutation) was the least sensitive among FGFR2-mutated lines. [2] At a clinically achievable concentration of 1 µmol/L, NVP-BGJ398 inhibited anchorage-independent growth (≥50% growth inhibition) in 3 out of 4 FGFR2-mutated cell lines, but in only 3 out of 11 (27%) cell lines with wild-type FGFR2, indicating a tighter correlation with FGFR2 mutation status. [2] NVP-BGJ398 inhibited FGFR2 pathway signaling in FGFR2-mutant cells. It blocked phosphorylation of FRS2α and ERK in a time-dependent manner in cells carrying the FGFR2 N550K mutation (AN3CA, MFE296) and blocked both basal and FGF7-stimulated phosphorylation in cells with the S252W mutation (MFE280). However, restoration of ERK signaling was observed within 24 to 72 hours. The inhibitor caused modest or variable effects on AKT phosphorylation. In FGFR2 wild-type cells (SNGM, HEC1A), NVP-BGJ398 treatment did not alter ERK or AKT signaling. [2] Treatment with NVP-BGJ398 induced a significant increase in the fraction of cells in G0–G1 phase arrest and a significant increase in apoptosis in FGFR2-mutated cell lines (AN3CA, MFE296, MFE280), but had minimal to no effect on cell cycle or apoptosis in FGFR2 wild-type cell lines (SNGM, HEC1A). [2] |
| ln Vivo |
Infigratinib is given to athymic nude mice that have had RT112/luc1 tumors implanted subcutaneously. The administration options include an intravenous bolus of 5 mg/kg in NMP/PEG200 (1:9, v/v) or an oral gavage of a suspension in PEG300/D5W (2:1, v/v) at a dose of 20 mg/kg. According to the pertinent pharmacokinetic (PK) parameters, infigratinib has a 32% oral bioavailability in this investigation. Following intravenous administration, infigratinib exhibits a high volume of distribution (26 L/kg) due to its quick distribution from the vascular compartment into the peripheral tissues. With a clearance of 3.3 L/h/kg (61% of liver blood flow), the plasma level is high. Based on AUC, the ratio of tumor to plasma following oral dosing is found to be 10[1]. The growth of endometrial cancer xenograft models with FGFR2 mutations is markedly inhibited by infigratinib (30 mg/kg)[2]. In mouse xenograft models derived from FGFR2-mutated endometrial cancer cells (AN3CA, MFE296), daily oral administration of NVP-BGJ398 at 30 mg/kg significantly delayed tumor growth. [2] In a long-term study using the FGFR2 wild-type SNGM xenograft model, NVP-BGJ398 showed no inhibitory effects. However, in the FGFR2 wild-type HEC1A xenograft model, NVP-BGJ398 treatment did delay tumor growth, despite showing no in vitro efficacy in these cells. [2] |
| Enzyme Assay | The purified GST-fusion FGFR3-K650E kinase domain phosphorylates a synthetic substrate in the presence of radiolabeled ATP to measure the enzymatic kinase activity. Enzyme activities are determined by combining 10 μL of the corresponding substrate mixture (peptidic substrate, ATP, and [γ33P]ATP) with 10 μL of a 3-fold concentrated Infigratinib solution or control. The assay buffer is mixed with 10 μL of a concentrated enzyme solution three times over to start the reactions. The following are the assay components' final concentrations: 0.5 μM ATP (γ-[33P]-ATP 0.1 μCi), 3 mM MnCl2, 3 mM MgCl2, 1 mM DTT, 250 μg/mL PEG 20000, 2 μg/mL poly(EY) 4:1, 1% DMSO, and 10 ng of GST-FGFR3-K650E were added. The assay is performed using the filter binding (FB) method in 96-well plates for 10 minutes at room temperature and 30 μL of final volume, which includes the components mentioned above. The following measurement is used to determine the amount of 33P incorporated into the polypeptidic substrates after 20 μL of 125 mM EDTA is added to stop the enzymatic reactions: Using a disconnected vacuum source, mount the Immobilon-PVDF membranes on a vacuum manifold and transfer 30 μL of the stopped reaction mixture onto them after they have been soaked in methanol for 5 minutes, rinsed with water, and soaked in 0.5% H3PO4 for 5 minutes. Following spotting, each well is rinsed with 200 μL of 0.5% H3PO4 and vacuumed. After extracting the free membranes, they are shaker-washed four times with 1% H3PO4 and once with ethanol. After desiccating the membranes, 10 μL of a scintillation fluid are added per well. In the end, the plates are sealed and counted using a microplate scintillation counter. By using linear regression analysis to determine the percentage inhibition of NVP-BGJ398[1], IC50 values are determined. |
| Cell Assay |
RPMI-1640 medium supplemented with 10% FBS, 4.5 g/L glucose, 1.5 g/L sodium bicarbonate, and Pen/Strep is used to cultivate mouse BaF3 cell lines. Twice a week, cells are passed through. A Luciferase bioluminescent assay is used to evaluate compound-mediated inhibition of BaF3 cell proliferation and viability. Using a μFill liquid dispenser, exponentially growing BaF3 or BaF3 Tel-TK cells are seeded at 50 μL/well into 384-well plates (4250 cells/well) in fresh medium. After being serially diluted in DMSO, infigratinib is arranged in a 384-well polypropylene plate. The plates were then incubated at 37°C (5% CO2) for 48 hours after 50 nL of the compound was transferred into them using the pintool transfer device. Next, add 25 μL of Bright-Glo, and use an Analyst-GT to measure the luminescence. A logistic fit of the percent cell viability as a function of the logarithm of inhibitor concentration is generated using specialized curve-fitting software. The concentration of a compound required to lower cell viability to 50% of a DMSO control is known as the IC50 value[1]. Proliferation Assay: Cells were plated in 24-well plates at a density of 2×10^5 to 5×10^5 and treated with increasing concentrations of NVP-BGJ398 (0.001 to 10 µmol/L). After 7 days, cells were harvested by trypsinization and counted. Growth inhibition was calculated, and IC50 values were interpolated from dose-response curves. [2] Soft Agar Clonogenic Assay: A bottom layer of 0.5% agar was placed in 24-well plates. Cells (5×10^3) were mixed into a top layer of 0.3% agar prepared with or without 1 µmol/L NVP-BGJ398 and plated in quadruplicate. Plates were incubated for up to 5 weeks. Colonies were stained and counted visually. Percent inhibition was calculated versus untreated controls. [2] Western Blot Analysis: Cells were treated with NVP-BGJ398, washed, and lysed. Protein concentrations were determined. Proteins were resolved by SDS-PAGE, transferred to membranes, and probed with antibodies against phospho-FRS2α, phospho-AKT, phospho-ERK, and α-Tubulin. For MFE280 cells, stimulation with FGF7 (30 ng/mL for 30 minutes) was performed prior to inhibitor treatment. Detection was performed using chemifluorescence. [2] Cell Cycle Analysis: Cells in log phase were treated with NVP-BGJ398 for 72 hours. Cells were stained and analyzed by flow cytometry to determine the fraction in different cell cycle phases. [2] Apoptosis Assay (Annexin V/PI): Cells were treated with NVP-BGJ398 for 72 hours. Apoptosis was detected using Annexin V-FITC and propidium iodide staining followed by flow cytometry. [2] |
| Animal Protocol |
Mice: HsdNpa female: Athymic Nude-nu mice are employed. Infigratinib is taken orally for 12 straight days at doses of 10 and 30 mg/kg/qd. It is prepared as a suspension in PEG300/D5W (2:1, v/v). ANOVA is used to analyze the tumor and body weight data, and Dunnett's test is used to compare the treatment group to the control group post hoc. For intragroup comparison, the post hoc Tukey test is employed. To perform statistical analysis, use GraphPad Prism 4.02. One calculates the T/C (%) value as a measure of efficacy. Rats: Woman in the nude 6 to 9-week-old Rowett rats are utilized. The tumor-bearing rats (n=8) receive infigratinib intraperitoneally (gavage) once a day for 20 days at doses of 5, 10, and 15 mg/kg/qd (free base equivalents). The drug is prepared as a solution in acetic acid-acetate buffer pH 4.6/PEG300 (1:1, v/v). It uses five milliliters per kilogram. The formula for determining tumor volumes is length×width×height×π/6, which can be measured using calipers. Antitumor activity is represented as T/C (%), which is calculated as (mean change in tumor volume of treated animals / mean change in tumor volume of control animals)×100. One calculates regressions (%). Female athymic mice (4-6 weeks old) were injected subcutaneously in the flank with endometrial cancer cells (2×10^7 cells per mouse). After tumors reached a mean volume of approximately 105 mm³ (7 days), mice were stratified into treatment groups based on tumor volume and weight. Mice (10 per group) were treated daily via oral gavage with: (i) vehicle control, or (ii) NVP-BGJ398 at 30 mg/kg (formulated as 6 mg in 0.5 mL PEG300 and 0.5 mL acetic acid/acetate buffer, pH 4.68). Tumor volumes were measured serially. [2] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Absorption Mean (%CV) Cmax is 282.5 ng/mL (54%) and AUC0-24h is 3780 ngxh/mL (59%) for infigratinib. Infigratinib Cmax and AUC increase more than proportionally across the dose range of 5 to 150 mg and steady state is achieved within 15 days. At steady state, median time to achieve peak infigratinib plasma concentration (Tmax) is six hours, with a range between two and seven hours. Mean (%CV) Cmax is 42.1 ng/mL (65%) for BHS697 and 15.7 ng/mL (92%) for CQM157. Mean (%CV) AUC0-24h is 717 ngxh/mL (55%) for BHS697 and 428 ngxh/mL (72%) for CQM157. In healthy subjects, a high-fat and high-calorie meal increased AUCinf of infigratinib by 80%-120% and Cmax by 60%-80%. The median Tmax also shifted from four hours to six hours. A low-fat low-calorie meal increased the mean AUCinf of infigratinib by 70% and Cmax by 90%/ Route of Elimination Following administration of a single oral dose of radiolabeled infigratinib in healthy subjects, approximately 77% of the dose was recovered in feces, where 3.4% of the dose was in the unchanged parent form. About 7.2% was recovered in urine with 1.9% of the dose was unchanged. Volume of Distribution At steady state, the geometric mean (CV%) apparent volume of distribution of infigratinib was 1600 L (33%). In rats receiving a single oral dose, infigratinib had brain-to-plasma concentration ratios (based on AUC0-inf) of 0.682. Clearance The geometric mean (CV%) total apparent clearance (CL/F) of infigratinib was 33.1 L/h (59%) at steady state. Metabolism / Metabolites According to _in vitro_ findings, about 94% of infigratinib is metabolized by CYP3A4 and about 6% of the drug is metabolized by flavin-containing monooxygenase 3 (FMO3). About 38% of the dose is circulating parent drug in the plasma and BHS697 and CQM157 are two major metabolites of infigratinib that are each found at >10% of the dose. They are pharmacologically active, with BHS697 representing about 16% to 33% of the overall pharmacological activity of infigratinib and CQM157 contributing to about 9% to 12%. BHS697 undergoes further metabolism mediated by CYP3A4 and CQM157 is metabolized through both Phase I and Phase II biotransformation pathways. The exact metabolic pathways and the structure of BHS697 and CQM157 are not fully characterized. Biological Half-Life The geometric mean (CV%) terminal half-life of infigratinib was 33.5 h (39%) at steady state. |
| Toxicity/Toxicokinetics |
Hepatotoxicity In the open label clinical trials of infigratinib for advanced or metastatic cholangiocarcinoma, adverse events were common and led to dose interruptions in 64%, dose reductions in 60% of patients, and permanent discontinuations in 15% largely for hyperphosphatemia, infections and sepsis rather than liver injury. In preregistration trials in 108 patients, ALT elevations arose in 51% and to above 5 times ULN in 6%. The elevations were typically self-limited and resolved rapidly with or without dose adjustments. No patients developed clinically apparent liver injury or jaundice. Since its approval, there have been no reports clinically apparent liver injury attributed to infigratinib. However, the total clinical experience with its use has been limited and the frequency of serum aminotransferase elevations during therapy suggest that clinically significant liver injury may occur. Likelihood score: E* (unproven but possible, rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Infigratinib is no longer marketed in the US. No information is available on the clinical use of infigratinib during breastfeeding. Because infigratinib is 96.8% bound to plasma proteins, the amount in milk is likely to be low. However, because of its potential toxicity in the breastfed infant and its half-life of 33.5 hours, the manufacturer recommends that breastfeeding be discontinued during infigratinib therapy and for 1 month after the last dose. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Infigratinib is about 96.8% bound to plasma proteins, primarily to lipoprotein. Protein binding is concentration-dependent. |
| References |
[1]. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J Med Chem . 2011 Oct 27;54(20):7066-83. [2]. Activity of the fibroblast growth factor receptor inhibitors dovitinib (TKI258) and NVP-BGJ398 in human endometrial cancer cells. Mol Cancer Ther. 2013 May;12(5):632-42. |
| Additional Infomation |
Infigratinib Phosphate is the phosphate salt form of infigratinib, an orally bioavailable pan-inhibitor of human fibroblast growth factor receptors (FGFRs) with potential antiangiogenic and antineoplastic activities. Upon administration, infigratinib selectively binds to and inhibits the activities of FGFRs, which may result in the inhibition of angiogenesis and cell proliferation, and the induction of cell death in tumors with activating FGFR amplifications, mutations, or fusions. FGFRs are a family of receptor tyrosine kinases that are involved in tumor cell differentiation and proliferation, tumor angiogenesis, and tumor cell survival. Activating FGFR amplifications, mutations, or fusions occur in various cancer cell types. See also: Infigratinib (has active moiety). A novel series of N-aryl-N'-pyrimidin-4-yl ureas has been optimized to afford potent and selective inhibitors of the fibroblast growth factor receptor tyrosine kinases 1, 2, and 3 by rationally designing the substitution pattern of the aryl ring. On the basis of its in vitro profile, compound 1h (NVP-BGJ398) was selected for in vivo evaluation and showed significant antitumor activity in RT112 bladder cancer xenografts models overexpressing wild-type FGFR3. These results support the potential therapeutic use of 1h as a new anticancer agent.[1] The recent identification of activating fibroblast growth factor receptor 2 (FGFR2) mutations in endometrial cancer has generated an opportunity for a novel target-based therapy. Here, we explore the therapeutic potential of 2 FGFR inhibitors, the multikinase inhibitor dovitinib (TKI258) and the more selective FGFR inhibitor NVP-BGJ398 for the treatment of endometrial cancer. We examined the effects of both inhibitors on tumor cell growth, FGFR2 signaling, cell cycle, and apoptosis using a panel of 20 molecularly characterized human endometrial cancer cell lines. Anchorage-independent growth was studied using soft agar assays. In vivo studies were conducted using endometrial cancer xenograft models. Cell lines with activating FGFR2 mutations (S252W, N550K) were more sensitive to dovitinib or NVP-BGJ398 when compared with their FGFR2 wild-type counterparts (P = 0.073 and P = 0.021, respectively). Both agents inhibited FGFR2 signaling, induced cell-cycle arrest, and significantly increased apoptosis in FGFR2-mutant lines. In vitro, dovitinib and NVP-BGJ398 were both potent at inhibiting cell growth of FGFR2-mutant endometrial cancer cells, but the activity of dovitinib was less restricted to FGFR2-mutant lines when compared with NVP-BGJ398. In vivo, dovitinib and NVP-BGJ398 significantly inhibited the growth of FGFR2-mutated endometrial cancer xenograft models. In addition, dovitinib showed significant antitumor activity in FGFR2 wild-type endometrial cancer xenograft models including complete tumor regressions in a long-term in vivo study. Dovitinib and NVP-BGJ398 warrant further clinical evaluation in patients with FGFR2-mutated endometrial cancer. Dovitinib may have antitumor activity in endometrial cancer beyond FGFR2-mutated cases and may permit greater flexibility in patient selection.[2] NVP-BGJ398 is a highly selective, pan-FGFR kinase inhibitor, optimized through rational drug design. Its selectivity suggests a potentially more favorable toxicity profile compared to non-selective multi-kinase inhibitors. [2] Activating FGFR2 mutations (e.g., S252W in ligand-binding domain, N550K in kinase domain) are found in a subset of endometrial cancers and confer sensitivity to FGFR inhibition. The frequency of these mutations might be higher in recurrent/metastatic disease compared to primary tumors. [2] The preclinical data from this study supported the rationale for clinical trials of NVP-BGJ398 in patients with recurrent endometrial cancer harboring FGFR2 mutations. [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
|
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.5187 mL | 7.5934 mL | 15.1867 mL | |
| 5 mM | 0.3037 mL | 1.5187 mL | 3.0373 mL | |
| 10 mM | 0.1519 mL | 0.7593 mL | 1.5187 mL |