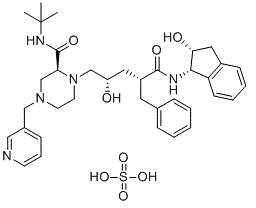
Indinavir sulfate (IDV; formerly known as MK-639; DRG-0233; L735524; trade name Crixivan), the sulfate salt of indinavir, is a potent and specific HIV protease inhibitor with antiviral effects and good oral bioavailability. Indinavir is used as a component of highly active antiretroviral therapy to treat HIV/AIDS. It is soluble white powder administered orally in combination with other antiviral drugs. The drug prevents protease from functioning normally. Consequently, HIV viruses cannot reproduce, causing a decrease in the viral load. Commercially sold indinavir is indinavir anhydrous, which is indinavir with an additional amine in the hydroxyethylene backbone. This enhances its solubility and oral bioavailability, making it easier for users to intake. It was synthetically produced for the purpose of inhibiting the protease in the HIV virus.
Physicochemical Properties
| Molecular Formula | C36H49N5O8S |
| Molecular Weight | 711.87 |
| Exact Mass | 711.33 |
| Elemental Analysis | C, 60.74; H, 6.94; N, 9.84; O, 17.98; S, 4.50 |
| CAS # | 157810-81-6 |
| Related CAS # | Indinavir;150378-17-9;Indinavir sulfate ethanolate;2563866-80-6 |
| PubChem CID | 5462355 |
| Appearance | White to off-white solid powder |
| Boiling Point | 877.9ºC at 760 mmHg |
| Melting Point | 150-153ºC |
| Flash Point | 484.7ºC |
| Vapour Pressure | 5.56E-33mmHg at 25°C |
| LogP | 3.952 |
| Hydrogen Bond Donor Count | 6 |
| Hydrogen Bond Acceptor Count | 11 |
| Rotatable Bond Count | 12 |
| Heavy Atom Count | 50 |
| Complexity | 1030 |
| Defined Atom Stereocenter Count | 5 |
| SMILES | O=C([C@@H](C[C@H](O)CN(CCN(CC1=CN=CC=C1)C2)[C@@H]2C(NC(C)(C)C)=O)CC3=CC=CC=C3)N[C@H]4C(C=CC=C5)=C5C[C@H]4O.O=S(O)(O)=O |
| InChi Key | NUBQKPWHXMGDLP-BDEHJDMKSA-N |
| InChi Code | InChI=1S/C36H47N5O4.H2O4S/c1-36(2,3)39-35(45)31-24-40(22-26-12-9-15-37-21-26)16-17-41(31)23-29(42)19-28(18-25-10-5-4-6-11-25)34(44)38-33-30-14-8-7-13-27(30)20-32(33)43;1-5(2,3)4/h4-15,21,28-29,31-33,42-43H,16-20,22-24H2,1-3H3,(H,38,44)(H,39,45);(H2,1,2,3,4)/t28-,29+,31+,32-,33+;/m1./s1 |
| Chemical Name | (2S)-1-[(2S,4R)-4-benzyl-2-hydroxy-5-[[(1S,2R)-2-hydroxy-2,3-dihydro-1H-inden-1-yl]amino]-5-oxopentyl]-N-tert-butyl-4-(pyridin-3-ylmethyl)piperazine-2-carboxamide sulfate |
| Synonyms | trade name: Crixivan; DRG-0233; DRG0233; L-735 524 sulfate; DRG 0233; MK-639 sulfate; L 735 524; MK 639; L735 524; MK639; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | MMP-2;HIV-1 |
| ln Vitro |
Indinavir sulfate (0-50 μM; 18 h) inhibits the G0/G1 phase of the lymphocyte cell cycle in PBMCs and reduces the ability of the cells to proliferate lymphomegaly[1]. In vitro, indinavir sulfate (40 μM–40 nM; 5 days) inhibits Huh7 and SK-HEP-1 hepatocarcinoma cells' ability to invade cells and activate MMPs-2 (40 μM–40 nM; 48 h)[2]. |
| ln Vivo | Indinavir sulfate (70 mg/kg; i.g.; once a day for 3 weeks) inhibits the growth of hepatocarcinoma cells in vivo[2]. |
| Cell Assay |
Cell Line: PBMCs (from healthy and HIV-infected volunteers) Concentration: 0-50 µM Incubation Time: 18 h (pretreatment; stimulation with anti-CD3 for an additional 48 hours) Result: Blocked anti-CD3-induced cell-cycle progression in a dose-dependent manner. Resulted in dose-dependent reduction of lymphoproliferative responses. |
| Animal Protocol |
Animal Model: Nude mice(s.c. into Huh7 and SK-HEP-1 cells)[2]. Dosage: 70 mg/kg Administration: Oral gavage; once a day for 3 weeks. Result: Delaied the growth of s.c. implanted hepatocarcinoma xenografts in nude mice compared with placebo. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion In a study in HIV-infected children 4-17 years of age receiving an antiretroviral regimen that included oral indinavir (initial dosage of 500 mg/sq m every 8 hours; subsequent dosage averaging 2043 mg/sq m daily in 3 or 4 doses); peak and trough plasma concentrations averaging 7.3 and 0.29 ug/ml, respectively. Indinavir is rapidly absorbed after oral administration, with peak levels achieved in approximately 1 hour. Unlike other drugs in this class, food can adversely affect indinavir bioavailability; a high-calorie, high-fat meal reduces plasma concentrations by 75%. Indinavir is excreted principally in the feces, both as unabsorbed drug and metabolites. Following oral administration of 400 mg of radiolabeled indinavir, 83% of the dose is recovered in feces (19.1% as unchanged drug) and 19% is recovered in urine (9.4% as unchanged drug). Following oral administration of a single 700- or 1000-mg dose of indinavir, 10.4 or 12%, respectively, is excreted unchanged in urine. To characterize steady-state indinavir pharmacokinetics in cerebrospinal fluid and plasma, 8 adults infected with human immunodeficiency virus underwent intensive cerebrospinal fluid sampling while receiving indinavir (800 mg every 8 hours) plus nucleoside reverse transcriptase inhibitors. Nine and 11 serial cerebrospinal fluid and plasma samples, respectively, were obtained from each subject. Free indinavir accounted for 94.3% of the drug in cerebrospinal fluid and 41.7% in plasma. Mean values of cerebrospinal fluid peak concentration, concentration at 8 hours, and area under the concentration-time profile calculated over the interval 0 to 8 hours (AUC(0-8)) for free indinavir were 294 nmol/L, 122 nmol/L, and 1616 nmol/L x hr, respectively. The cerebrospinal fluid-to-plasma AUC(0-8) ratio for free indinavir was 14.7% +/- 2.6% and did not correlate with indexes of blood-brain barrier integrity or intrathecal immune activation. Indinavir achieves levels in cerebrospinal fluid that should contribute to control of human immunodeficiency virus type 1 replication in this compartment. The cerebrospinal fluid-to-plasma AUC (0-8) ratio suggests clearance mechanisms in addition to passive diffusion across the blood-cerebrospinal fluid barrier, perhaps by P-glycoprotein-mediated efflux. For more Absorption, Distribution and Excretion (Complete) data for INDINAVIR SULFATE (7 total), please visit the HSDB record page. Metabolism / Metabolites Indinavir is metabolized to at least 7 metabolites including 1 glucuronide conjugate and 6 oxidative metabolites. Major metabolic pathways identified include glucuronidation at the pyridine nitrogen, pyridine N-oxidation, para-hydroxylation of the phenylmethyl group, 3-hydroxylation of the indan, and N-depyridomethylation. In vitro studies indicate that cytochrome P-450 isoenzyme CYP3A4 is the major enzyme involved in the formation of the oxidative metabolites. Biological Half-Life In a study in adults with cirrhosis and mild to moderate hepatic impairment, the elimination half-life of the drug was prolonged to 2.8 hours. The plasma half-life of indinavir averages 1.8 hours. In HIV-infected children 4-17 years of age receiving an antiretroviral regimen that included oral indinavir, plasma half-life of the drug averaged 1.1 hours. |
| Toxicity/Toxicokinetics |
Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Indinavir is no longer marketed in the US. Published experience with indinavir during breastfeeding is limited, but some infants may achieve high levels of the drug in breastmilk. Indinavir is not a recommended agent during breastfeeding. Achieving and maintaining viral suppression with antiretroviral therapy decreases breastfeeding transmission risk to less than 1%, but not zero. Individuals with HIV who are on antiretroviral therapy with a sustained undetectable viral load and who choose to breastfeed should be supported in this decision. If a viral load is not suppressed, banked pasteurized donor milk or formula is recommended. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Gynecomastia has been reported among men receiving highly active antiretroviral therapy. Gynecomastia is unilateral initially, but progresses to bilateral in about half of cases. No alterations in serum prolactin were noted and spontaneous resolution usually occurred within one year, even with continuation of the regimen. Some case reports and in vitro studies have suggested that protease inhibitors might cause hyperprolactinemia and galactorrhea in some male patients, although this has been disputed. The relevance of these findings to nursing mothers is not known. The prolactin level in a mother with established lactation may not affect her ability to breastfeed. Interactions Studies have not been done with the cytochrome p450 CYP3A4 substrates astemizole, cisapride, midazolam, terfenadine, and triazolam; because competition for CYP3A4 by indinavir could result in inhibition of the metabolism of these medications and elevated plasma concentrations, there is a potential for serious and/or life-threatening side effects; concurrent use of indinavir with any of these medications is not recommended. Concurrent administration /of cimetidine and indinavir/ does not affect the area under the plasma concentration-time curve (AUC) of indinavir. Amprenavir interferes with the metabolism of rifabutin and significantly increases rifubutin serum concentrations; it is recommended that the dose of rifabutin be reduced by at least half of the recommended dose; rifabutin decreases the AUC of amprenavir by 15%; patients should be monitored for neutropenia once a week and as clinically indicated if rifabutin is given concurrently with amprenavir. Concurrent use /with clarithromycin/ results in a 29% increase in the AUC of indinavir and a 53% increase in the AUC of clarithromycin; dosing modification is not required. For more Interactions (Complete) data for INDINAVIR SULFATE (18 total), please visit the HSDB record page. Non-Human Toxicity Values LD50 Dog Intraperitoneal > 640 mg/kg LD50 Dog oral > 640 mg/kg LD50 Mouse Intraperitoneal > 5 g/kg LD50 Mouse oral > 5 g/kg For more Non-Human Toxicity Values (Complete) data for INDINAVIR SULFATE (6 total), please visit the HSDB record page. |
| References |
[1]. The HIV protease inhibitor Indinavir inhibits cell-cycle progression in vitro in lymphocytes of HIV-infected and uninfected individuals. Blood. 2001 Jul 15;98(2):383-9. [2]. Evaluation of antitumoral properties of the protease inhibitor indinavir in a murine model of hepatocarcinoma. Clin Cancer Res. 2006 Apr 15;12(8):2634-9. [3]. Kinetic, stability, and structural changes in high-resolution crystal structures of HIV-1 protease with drug-resistant mutations L24I, I50V, and G73S. J Mol Biol. 2005 Dec 9;354(4):789-800. [4]. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med Infect Dis. 2020 May-Jun;35:101646. [5]. AIDS.1996 May;10(5):485-92. [6]. J Mol Biol.2005 Dec 9;354(4):789-800. |
| Additional Infomation |
Therapeutic Uses HIV Protease Inhibitors. Indinavir with antiretroviral agents is indicated for the treatment of HIV infection. /Included in US product labeling/ Drug Warnings Nephrolithiasis/urolithiasis, which may present as flank pain with or without hematuria (including microscopic hematuria), has been reported in about 9% of adults and 29% of pediatric patients receiving indinavir. The most frequent adverse effects associated with indinavir therapy involve the GI tract. ...In treatment-naive HIV-infected adults, abdominal pain, nausea, vomiting, and diarrhea occurred in 16.6, 11.7, 8.4, and 3.3%, respectively, and acid regurgitation, anorexia, dyspepsia, increased appetite, and taste perversion occurred in 1.5-2.7% of patients receiving indinavir monotherapy. In patients in study 028 receiving indinavir in conjunction with zidovudine, abdominal pain, nausea, vomiting, and diarrhea occurred in 16, 31.9, 17.8, and 3%, respectively, and acid regurgitation, anorexia, dyspepsia, increased appetite, and taste perversion occurred in 1.5-8.4% of patients. ...Safety and efficacy of indinavir in pediatric patients have not been established. Indinavir has been used in a limited number of HIV-infected children 3 months of age or older without unusual adverse effects. However, nephrolithiasis/urolithiasis has been reported more frequently in pediatric patients receiving indinavir (29%) than in adults receiving the drug (9.2%). Asymptomatic hyperbilirubinemia (i.e., total serum bilirubin concentrations exceeding 2.5 mg/dL) has occurred in about 14% of patients receiving indinavir in clinical studies. Asymptomatic bilirubinemia usually has been reported as elevated indirect bilirubin and has been associated with increased serum AST (SGOT) or ALT (SGPT) concentrations only rarely (i.e., in less than 1% of patients receiving the drug). Acute hepatitis sometimes resulting in hepatic failure and death have been reported in a few patients receiving indinavir in conjunction with other drugs. ...Jaundice was reported in 1.5-2.1% of patients receiving indinavir. For more Drug Warnings (Complete) data for INDINAVIR SULFATE (21 total), please visit the HSDB record page. |
Solubility Data
| Solubility (In Vitro) |
DMSO : ~100 mg/mL ( ~140.47 mM ) H2O :~50 mg/mL (~70.24 mM ) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (3.51 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (3.51 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (3.51 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 10% DMSO+40% PEG300+5% Tween-80+45% Saline: ≥ 2.5 mg/mL (3.51 mM) Solubility in Formulation 5: 100 mg/mL (140.48 mM) in PBS (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.4048 mL | 7.0238 mL | 14.0475 mL | |
| 5 mM | 0.2810 mL | 1.4048 mL | 2.8095 mL | |
| 10 mM | 0.1405 mL | 0.7024 mL | 1.4048 mL |