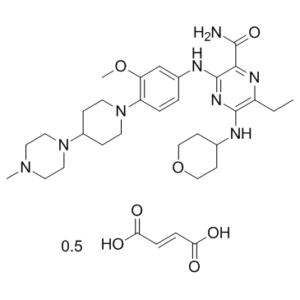
Gilteritinib hemifumarate (ASP2215; ASP-2215 hemifumarate; Xospata), the hemifumarate salt of Gilteritinib (ASP2215), is a marketed anti-AML drug acting as a dual inhibitor of FLT3/AXL with IC50 values of 0.29 nM and 0.73 nM for FLT3 and AXL respectively. The US FDA approved gelderitinib in November 2018 for the treatment of patients with relapsed or refractory acute myeloid leukemia (AML). With an IC50 roughly 800-fold higher than that needed to inhibit c-KIT (230 nM), it potently inhibits FLT3. Treatment options for leukemia like AML with FLT3-ITD and FLT3-D835 mutations include gelderitinib. With an IC50 value of 0.29 nM for FLT3, gelderitinib is approximately 800-fold more potent than c-KIT and inhibits FLT3, leukocyte tyrosine kinase (LTK), anaplastic lymphoma kinase (ALK), and AXL kinases by over 50% in vitro among the 78 tyrosine kinases tested.
Physicochemical Properties
| Molecular Formula | C29H44N8O3₀.₅C₄H₄O₄ |
| Molecular Weight | 610.75 |
| Exact Mass | 1220.718 |
| Elemental Analysis | C, 60.96; H, 7.59; N, 18.35; O, 13.10 |
| CAS # | 1254053-84-3 |
| Related CAS # | Gilteritinib;1254053-43-4 |
| PubChem CID | 76970819 |
| Appearance | White to yellow solid powder |
| Hydrogen Bond Donor Count | 8 |
| Hydrogen Bond Acceptor Count | 24 |
| Rotatable Bond Count | 20 |
| Heavy Atom Count | 88 |
| Complexity | 904 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | CCC1=C(N=C(C(=N1)C(=O)N)NC2=CC(=C(C=C2)N3CCC(CC3)N4CCN(CC4)C)OC)NC5CCOCC5.CCC1=C(N=C(C(=N1)C(=O)N)NC2=CC(=C(C=C2)N3CCC(CC3)N4CCN(CC4)C)OC)NC5CCOCC5.C(=C/C(=O)O)\C(=O)O |
| InChi Key | UJOUWHLYTQFUCU-WXXKFALUSA-N |
| InChi Code | InChI=1S/2C29H44N8O3.C4H4O4/c2*1-4-23-28(31-20-9-17-40-18-10-20)34-29(26(33-23)27(30)38)32-21-5-6-24(25(19-21)39-3)37-11-7-22(8-12-37)36-15-13-35(2)14-16-36;5-3(6)1-2-4(7)8/h2*5-6,19-20,22H,4,7-18H2,1-3H3,(H2,30,38)(H2,31,32,34);1-2H,(H,5,6)(H,7,8)/b;;2-1+ |
| Chemical Name | (E)-but-2-enedioic acid;6-ethyl-3-[3-methoxy-4-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]anilino]-5-(oxan-4-ylamino)pyrazine-2-carboxamide |
| Synonyms | ASP2215 hemifumarate; ASP-2215 hemifumarate; ASP 2215 hemifumarate; Gilteritinib hemifumarate; Trade name: Xospata |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
FLT3 (IC50 = 0.29 nM); LTK (IC50 = 0.35 nM); AXL (IC50 = 0.73 nM); EML4-ALK (IC50 = 1.2 nM); c-KIT (IC50 = 230 nM)
The targets of Gilteritinib hemifumarate (active ingredient: Gilteritinib/ASP2215) are Fms-like tyrosine kinase 3 (FLT3) and AXL receptor tyrosine kinase; it is a type I tyrosine kinase inhibitor with high selectivity for FLT3 and AXL, and weak activity against c-KIT. [2] Gilteritinib hemifumarate inhibits mutated forms of FLT3 including internal tandem duplication (FLT3-ITD), FLT3-D835Y point mutation, and FLT3-F691 mutation (weaker inhibitory activity against FLT3-F691); [2] |
| ln Vitro |
Gilteritinib (ASP2215), out of the 78 tyrosine kinases examined, inhibits the kinases FLT3, leukocyte tyrosine kinase (LTK), anaplastic lymphoma kinase (ALK), and AXL by more than 50% at 1 nM. For FLT3, the IC50 value is 0.29 nM, which makes it about 800-fold more potent than c-KIT[1]. Gilteritinib, at concentrations of either 1 nM (FLT3, LTK, ALK, and AXL) or 5 nM (TRKA, ROS, RET, and MER), inhibits the activity of eight of the 78 tested kinases by more than 50%. For FLT3, the IC50 is 0.29 nM, and for AXL, it is 0.73 nM. At an IC50 that is roughly 800-fold more potent than that needed to inhibit c-KIT (230 nM), gelderitinib inhibits FLT3. Gilteritinib's antiproliferative efficacy is assessed in relation to MV4-11 and MOLM-13 cells, which are endogenously FLT3-ITD-expressing. Gilteritinib inhibits the growth of MOLM-13 and MV4-11 cells after five days of treatment, with mean IC50s of 2.9 nM (95% CI: 1.4-5.8 nM) and 0.92 nM (95% CI: 0.23-3.6 nM), respectively. MV4-11 cell growth suppression is correlated with inhibition of FLT3 phosphorylation. Following two hours of treatment with 0.1 nM, 1 nM, and 10 nM Gilteritinib, respectively, the phosphorylated FLT3 levels are 57%, 8%, and 1% higher than those of the vehicle control cells. Furthermore, the suppression of phosphorylated ERK, STAT5, and AKT—all downstream targets of FLT3 activation—occurs at concentrations as low as 0.1 nM or 1 nM. Gilteritinib is administered to MV4-11 cells that expressed exogenous AXL in order to study its effects on AXL inhibition. Gilteritinib treatment reduces phosphorylated AXL levels by 38%, 29%, and 22% at concentrations of 1 nM, 10 nM, and 100 nM for 4 hours[2]. 1. Anti-proliferative activity in AML cell lines: The active component Gilteritinib from Gilteritinib hemifumarate was tested on MV4-11 and MOLM-13 AML cell lines (harboring FLT3 mutations) as well as Ba/F3 cells expressing mutated FLT3 (FLT3-ITD, FLT3-D835Y, FLT3-ITD-D835Y). MV4-11 cells were treated with DMSO or increasing concentrations of gilteritinib for 5 days, and cell viability was measured using CellTiter-Glo; the results showed dose-dependent inhibition of cell growth (mean ± SEM, quadruplicate experiments, representative result from three independent experiments). MOLM-13 cells were treated in the same manner, with consistent dose-dependent growth inhibition observed [2] 2. Inhibition of FLT3 phosphorylation and downstream signaling: MV4-11 cells were treated with DMSO or increasing concentrations of Gilteritinib (from Gilteritinib hemifumarate) for 2 hours. Immunoprecipitation and immunoblot analysis revealed dose-dependent reduction in phosphorylated FLT3 levels (densitometry values provided below blots, triplicate experiments). Additionally, immunoblot analysis showed that gilteritinib decreased the phosphorylation levels of FLT3 downstream targets (AKT, ERK, STAT5) in MV4-11 cells (triplicate experiments) [2] 3. Inhibitory activity against different FLT3 mutations: Gilteritinib (active component of Gilteritinib hemifumarate) exhibited potent inhibitory activity against FLT3-ITD and FLT3-D835Y mutations in cellular assays, and weaker activity against FLT3-F691 mutations [2] |
| ln Vivo |
After administering oral Gilteritinib at a dose of 10 mg/kg for four days, the concentration of the drug in tumors in MV4-11 xenografted mice was found to be over 20 times higher than that in plasma. MV4-11 tumor growth is dose-dependently inhibited during a 28-day treatment with gelderitinib, and at doses greater than 6 mg/kg, total tumor regression is induced. Furthermore, gerteritinib prolongs the survival of mice receiving intravenous MV4-11 cell transplants and reduces the tumor burden in the bone marrow[1]. 1. Plasma and tumor distribution: Nude mice subcutaneously xenografted with MV4-11 cells were administered a single oral dose of Gilteritinib (from Gilteritinib hemifumarate) at 1 mg/kg, 6 mg/kg, or 10 mg/kg. Plasma and tumor concentrations of gilteritinib were determined by high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS), with high intratumor distribution observed (mean ± SD: 2-3 animals for plasma, 3 animals for tumor) [2] 2. Inhibition of FLT3/STAT5 phosphorylation in xenograft tumors: Male mice xenografted with MV4-11 cells were orally treated with vehicle control or increasing concentrations of Gilteritinib (from Gilteritinib hemifumarate). Over a 24-hour time course, tumor protein lysates were collected to measure: (1) the ratio of phospho-FLT3 to total FLT3 by ELISA (normalized to vehicle control); (2) the ratio of phospho-STAT5 to total STAT5 by immunoblot (normalized to vehicle control) [2] 3. Antitumor activity in MV4-11 xenograft model: Male mice xenografted with MV4-11 cells were orally treated with vehicle control or Gilteritinib (from Gilteritinib hemifumarate) at 1 mg/kg, 3 mg/kg, 6 mg/kg, or 10 mg/kg once daily for 28 days. Gilteritinib dose-dependently reduced tumor volume (mean ± SEM, n=6 mice/group; P < 0.05, P < 0.01, P < 0.001 vs control on Day 28, Dunnett’s test), with no significant changes in body weight [2] 4. Tumor regression in FLT3 mutant xenograft models: Male nude mice xenografted with Ba/F3 cells expressing FLT3-ITD, FLT3-D835Y, or FLT3-ITD-D835Y were treated with 10 mg/kg or 30 mg/kg of oral Gilteritinib (from Gilteritinib hemifumarate) once daily for up to 7 days after confirmed tumor growth. Gilteritinib induced significant tumor regression in all three models (mean ± SEM, n=5 mice/group; P < 0.01, P < 0.001 vs control on Day 7, Dunnett’s test) [2] 5. Reduction of leukemic burden and improved survival: Female NOD-SCID mice engrafted with MV4-11-luc cells were treated with 30 mg/kg of oral Gilteritinib (from Gilteritinib hemifumarate) once daily for 56 days starting on Day 15. Whole-body imaging showed significant reduction in bone marrow infiltration of MV4-11-luc cells (mean ± SEM, n=10 mice/group; P < 0.001 vs control on Day 42, Student’s t-test). Kaplan–Meier analysis confirmed improved survival in gilteritinib-treated mice (P < 0.001 vs control, Log-rank test) [2] |
| Enzyme Assay |
TK-ELISA or off-chip mobility shift assays are used to test the kinase inhibitory activity of Gilteritinib against a panel of 78 tested kinases, with ATP concentrations roughly equivalent to each kinase's Km value. Initially, the inhibitory effect of each compound on TK activity is evaluated at two concentrations of Gilteritinib (1 nM and 5 nM). Next, additional research employing a range of Gilteritinib doses is carried out to ascertain IC50 values for c-KIT and kinases whose activity is inhibited by more than half at 1 nM Gilteritinib. FLT3, LTK, AXL, and c-KIT IC50 studies are carried out using TK-ELISA and MSA assays; the HTRF KinEASE-TK assay is used to determine the IC50 value of echinoderm microtubule-associated protein-like 4-ALK (EML4-ALK)[2]. Kinase selectivity assays were performed to evaluate Gilteritinib (active component of Gilteritinib hemifumarate). Purified FLT3, AXL, c-KIT, and other tyrosine kinases were incubated with gilteritinib in the presence of ATP and specific substrates (substrate details not provided) under optimized reaction conditions (temperature and incubation time not specified). Kinase activity was measured using an unspecified detection method to assess the inhibitory effect of gilteritinib on each kinase. The results confirmed high selectivity for FLT3 and AXL, with weak activity against c-KIT; [2] |
| Cell Assay |
The CellTiter-Glo Luminescent Cell Viability Assay is used to evaluate the impact of gelderitinib on MV4-11 and MOLM-13 cells. Further research is done to investigate the impact of quizartinib and gilteritinib on Ba/F3 cells that express FLT3-ITD, FLT3-D835Y, FLT3-ITD-D835Y, FLT3-ITD-F691 L, or FLT3-ITD-F691I. For five days, DMSO or increasing concentrations of Gilteritinib (0.01, 0.1, 1, 10, and 100 nM) are applied to MV4-11 and MOLM-13 cells. CellTiter-Glo is used to measure the viability of the cells[2]. 1. AML cell viability assay: - Step 1: MV4-11 and MOLM-13 AML cells were seeded in appropriate culture plates at an unspecified density and cultured under standard conditions to ensure logarithmic growth. - Step 2: Cells were treated with DMSO (control) or serial concentrations of Gilteritinib (from Gilteritinib hemifumarate) and incubated for 5 days. - Step 3: Cell viability was quantified using the CellTiter-Glo assay (detection principle not specified), with data collected in quadruplicate and presented as mean ± SEM. The experiment was repeated three times, with a representative result reported [2] 2. Immunoprecipitation and immunoblot for FLT3 phosphorylation: - Step 1: MV4-11 cells were cultured to the desired confluency and treated with DMSO or increasing concentrations of Gilteritinib (from Gilteritinib hemifumarate) for 2 hours. - Step 2: Cells were harvested and lysed with an unspecified lysis buffer; protein concentration was determined by an unspecified method. - Step 3: FLT3 protein was immunoprecipitated using a FLT3-specific antibody (details not provided), and immunoprecipitates were separated by SDS-PAGE (gel concentration not specified). - Step 4: Separated proteins were transferred to an unspecified membrane, probed with primary antibodies against phosphorylated and total FLT3, followed by secondary antibody incubation. - Step 5: Protein bands were detected by an unspecified method, and densitometry analysis was performed to quantify band intensities (values listed below blots). The experiment was conducted in triplicate [2] 3. Immunoblot for FLT3 downstream signaling proteins: - Step 1: MV4-11 cells were treated with DMSO or increasing concentrations of Gilteritinib (from Gilteritinib hemifumarate) for 2 hours as described above. - Step 2: Cell lysates were prepared, and equal amounts of protein were separated by SDS-PAGE, transferred to a membrane, and probed with primary antibodies against phosphorylated AKT, ERK, STAT5, and their total forms (loading control not specified). - Step 3: Protein bands were detected and analyzed (triplicate experiments) [2] |
| Animal Protocol |
Mice: In nude mice transplanted with MV4-11 AML cells, antitumor activity is assessed. Additionally, the pharmacokinetics in xenografted mice are examined. Gilteritinib (10 mg/kg) is given orally to MV4-11 xenografted mice for a period of four days as part of their treatment. Gilteritinib treatment for 28 days causes total tumor regression at doses greater than 6 mg/kg and dose-dependent inhibition of MV4-11 tumor growth[1]. 1. MV4-11 xenograft model (pharmacokinetic analysis): - Step 1: MV4-11 AML cells were subcutaneously injected into nude mice (gender/age not specified) at an unspecified cell number to establish xenograft models. - Step 2: When tumors reached an unspecified volume, mice were given a single oral dose of Gilteritinib (from Gilteritinib hemifumarate) at 1 mg/kg, 6 mg/kg, or 10 mg/kg (drug formulation/vehicle not specified). - Step 3: Blood and tumor samples were collected at unspecified time points after administration. - Step 4: Plasma and tumor concentrations of gilteritinib were measured by HPLC-MS/MS, with data presented as mean ± SD (2-3 animals for plasma, 3 animals for tumor) [2] 2. MV4-11 xenograft model (pharmacodynamic/efficacy analysis): - Step 1: MV4-11 cells were subcutaneously implanted into male nude mice (n=6 per group) to establish xenograft models. - Step 2: After tumor establishment (unspecified volume), mice were orally administered vehicle control or Gilteritinib (from Gilteritinib hemifumarate) at 1 mg/kg, 3 mg/kg, 6 mg/kg, or 10 mg/kg once daily for 28 days (drug formulation not specified). - Step 3: For pharmacodynamic analysis: Tumor tissues were collected over a 24-hour time course, protein lysates were prepared, and the ratios of phospho-FLT3/total FLT3 (ELISA) and phospho-STAT5/total STAT5 (immunoblot) were determined. - Step 4: For efficacy analysis: Tumor volume and body weight were measured at unspecified intervals, with statistical analysis (Dunnett’s test) performed on Day 28 [2] 3. Ba/F3-FLT3 mutant xenograft model: - Step 1: Ba/F3 cells expressing FLT3-ITD, FLT3-D835Y, or FLT3-ITD-D835Y were subcutaneously injected into male nude mice (n=5 per group) to establish xenograft models. - Step 2: After confirmed tumor growth (unspecified volume), mice were orally administered vehicle control, 10 mg/kg, or 30 mg/kg of Gilteritinib (from Gilteritinib hemifumarate) once daily for up to 7 days (drug formulation not specified). - Step 3: Tumor volume was measured at unspecified time points, with statistical analysis (Dunnett’s test) performed on Day 7 [2] 4. Intra-bone marrow transplantation model: - Step 1: MV4-11-luc cells (luciferase-expressing MV4-11 cells) were engrafted into female NOD-SCID mice via intra-bone marrow injection (injection site/cell number not specified). - Step 2: Starting on Day 15 after engraftment, mice (n=10 per group) were orally administered vehicle control or 30 mg/kg of Gilteritinib (from Gilteritinib hemifumarate) once daily for 56 days (drug formulation not specified). - Step 3: Whole-body bioluminescence imaging was performed at unspecified time points (e.g., Day 21, Day 42) to monitor bone marrow infiltration of MV4-11-luc cells (imaging parameters not specified). - Step 4: Mouse survival was recorded, and Kaplan–Meier analysis (Log-rank test) was conducted [2] |
| ADME/Pharmacokinetics |
1. Absorption and distribution: Gilteritinib (active component of Gilteritinib hemifumarate) was rapidly absorbed after oral administration to nude mice bearing MV4-11 xenografts, with high distribution in tumor tissues. Plasma and tumor concentrations were measured by HPLC-MS/MS after a single oral dose (1 mg/kg, 6 mg/kg, 10 mg/kg) [2] 2. Metabolism, excretion, half-life, and bioavailability: No information about the metabolism, excretion, half-life, or oral bioavailability of Gilteritinib hemifumarate was provided [2] |
| Toxicity/Toxicokinetics |
1. In vivo toxicity: No overt toxicity was observed in mouse models treated with Gilteritinib (from Gilteritinib hemifumarate). In the MV4-11 xenograft model, 28-day treatment with gilteritinib (1-10 mg/kg once daily) did not cause significant changes in mouse body weight, indicating good tolerability [2] |
| References |
[1]. ASP2215, a novel FLT3/AXL inhibitor: Preclinical evaluation in acute myeloid leukemia (AML). 2014 ASCO Annual Meeting. [2]. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Invest New Drugs. 2017 Oct;35(5):556-565. |
| Additional Infomation |
Gilteritinib Fumarate is the fumarate salt form of gilteritinib, an orally bioavailable inhibitor of the receptor tyrosine kinases (RTKs) FMS-like tyrosine kinase 3 (FLT3; STK1; FLK2), AXL (UFO; JTK11), anaplastic lymphoma kinase (ALK; CD246), and leukocyte receptor tyrosine kinase (LTK), with potential antineoplastic activity. Upon administration, gilteritinib binds to and inhibits both the wild-type and mutated forms of FLT3, AXL, ALK and LTK. This may result in an inhibition of FLT3-, AXL-, ALK-, and LTK-mediated signal transduction pathways and reduced proliferation in cancer cells that overexpress these RTKs. FLT3, AXL, ALK, and LTK, which are overexpressed or mutated in a variety of cancer cell types, play key roles in tumor cell growth and survival. See also: Gilteritinib (has active moiety). Drug Indication Xospata is indicated as monotherapy for the treatment of adult patients who have relapsed or refractory acute myeloid leukaemia (AML) with a FLT3 mutation. , 1. Background and composition: Gilteritinib hemifumarate is a salt form of Gilteritinib (ASP2215), a novel type I tyrosine kinase inhibitor with high selectivity for FLT3 and AXL. FLT3 is one of the most frequently mutated genes in acute myeloid leukemia (AML), and FLT3 mutations are associated with poor overall survival; AXL is involved in FLT3 activation and AML pathogenesis [2] 2. Mechanism of action: The active component Gilteritinib in Gilteritinib hemifumarate exerts antileukemic activity by inhibiting the phosphorylation of mutated FLT3 and its downstream signaling pathways (AKT, ERK, STAT5), thereby suppressing AML cell proliferation and inducing tumor regression in vivo [2] 3. Clinical potential: Preclinical studies demonstrated that Gilteritinib hemifumarate (via its active component Gilteritinib) has potent antileukemic activity in FLT3-mutated AML cell lines and mouse models (xenograft and intra-bone marrow transplantation models) with no overt toxicity, indicating it may be a promising next-generation FLT3 inhibitor for FLT3 mutation-positive AML [2] 4. Computational modeling: Computational modeling of Gilteritinib (active component of Gilteritinib hemifumarate) binding to wild-type FLT3 was performed. Gilteritinib was visualized as a ball-and-stick model (atoms colored by element type), and the protein surface was colored by electrostatic potential; the gatekeeper residue F691 was highlighted [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: 10 mg/mL (16.37 mM) in 50% PEG300 +50% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution; with sonication. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.6373 mL | 8.1867 mL | 16.3733 mL | |
| 5 mM | 0.3275 mL | 1.6373 mL | 3.2747 mL | |
| 10 mM | 0.1637 mL | 0.8187 mL | 1.6373 mL |