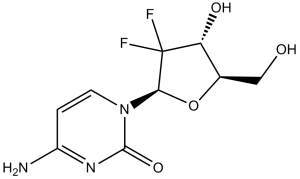
Gemcitabine (formerly LY-188011, NSC-613327; LY188011, NSC613327; dFdC; dFdCyd; trade name: Gemzar), an approved antimetabolite anticancer drug, is a potent DNA synthesis inhibitor with potential antineoplastic activity. With IC50s of 50 nM, 40 nM, 18 nM, and 12 nM, respectively, it suppresses the growth of PANC1, MIAPaCa2, BxPC3, and Capan2 cells. Difluorodeoxycytidine di- and triphosphate (dFdCDP, dFdCTP) are the active metabolites of gemcitabine that are produced intracellularly. The deoxynucleotide pool available for DNA synthesis is reduced when dFdCDP inhibits ribonucleotide reductase.
Physicochemical Properties
| Molecular Formula | C9H11F2N3O4 | |
| Molecular Weight | 263.2 | |
| Exact Mass | 263.071 | |
| Elemental Analysis | C, 41.07; H, 4.21; F, 14.44; N, 15.97; O, 24.31 | |
| CAS # | 95058-81-4 | |
| Related CAS # |
|
|
| PubChem CID | 60750 | |
| Appearance | White to off-white solid powder | |
| Density | 1.8±0.1 g/cm3 | |
| Boiling Point | 468.0±55.0 °C at 760 mmHg | |
| Melting Point | 168.64°C | |
| Flash Point | 236.8±31.5 °C | |
| Vapour Pressure | 0.0±2.6 mmHg at 25°C | |
| Index of Refraction | 1.652 | |
| LogP | -1.24 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 6 | |
| Rotatable Bond Count | 2 | |
| Heavy Atom Count | 18 | |
| Complexity | 426 | |
| Defined Atom Stereocenter Count | 3 | |
| SMILES | FC1([C@H](O)[C@@H](CO)O[C@H]1N1C=CC(N)=NC1=O)F |
|
| InChi Key | SDUQYLNIPVEERB-QPPQHZFASA-N | |
| InChi Code | InChI=1S/C9H11F2N3O4/c10-9(11)6(16)4(3-15)18-7(9)14-2-1-5(12)13-8(14)17/h1-2,4,6-7,15-16H,3H2,(H2,12,13,17)/t4-,6-,7-/m1/s1 | |
| Chemical Name | 4-amino-1-[(2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: This product requires protection from light (avoid light exposure) during transportation and storage. |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | DNA synthesis | |
| ln Vitro |
|
|
| ln Vivo |
|
|
| Cell Assay |
Proliferation assay. [5] The effect of curcumin on cell proliferation was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) uptake method as described previously. The cells (2,000 per well) were incubated with curcumin in triplicate in a 96-well plate and then incubated for 2, 4, or 6 days at 37°C. A MTT solution was added to each well and incubated for 2 h at 37°C. An extraction buffer (20% SDS and 50% dimethylformamide) was added, and the cells were incubated overnight at 37°C. The absorbance of the cell suspension was measured at 570 nm using an MRX Revelation 96-well multiscanner. This experiment was repeated twice, and the statistical analysis (simple linear regression analysis initially and then unpaired Student's t test that revealed significant differences between two sample means) was done to obtain the final values.[5] Apoptosis assay. [5] To determine whether curcumin can potentiate the apoptotic effects of gemcitabine in pancreatic cancer cells, we used a Live/Dead assay kit, which determines intracellular esterase activity and plasma membrane integrity. This assay uses calcein, a polyanionic, green fluorescent dye that is retained within live cells, and a red fluorescent ethidium bromide homodimer dye that can enter cells through damaged membranes and bind to nucleic acids but is excluded by the intact plasma membranes of live cells. Briefly, cells (5,000 per well) were incubated in chamber slides, pretreated with curcumin for 4 h, and treated with gemcitabine for 24 h. Cells were then stained with the assay reagents for 30 min at room temperature. Cell viability was determined under a fluorescence microscope by counting live (green) and dead (red) cells. This experiment was repeated twice and the statistical analysis was done. The values were initially subjected to one-way ANOVA, which revealed significant differences between groups, and then later compared among groups using unpaired Student's t test, which revealed significant differences between two sample means.[5] In a 96-well plate, BxPC-3, MIA PaCa-2, and PANC-1 cells are seeded. Cells are treated for a further 24 or 48 hours with vehicle, DMAPT, and/or Gemcitabine after 24 hours. Using the Cell Death Detection ELISA, apoptosis is measured in relation to vehicle-treated cells by counting the quantity of cytoplasmic histone-associated DNA fragments. |
|
| Animal Protocol |
Female BALB/c nude mice 5 mg/kg i.p. After 1 week of implantation, mice were randomized into the following treatment groups (n = 6) based on the bioluminescence measured after the first IVIS imaging: (a) untreated control (olive oil, 100 μL daily); (b) curcumin alone (1 g/kg), once daily p.o.; (c) gemcitabine alone (25 mg/kg), twice weekly by i.p. injection; and (d) combination of curcumin (1 g/kg), once daily p.o., and gemcitabine (25 mg/kg), twice weekly by i.p. injection. Tumor volumes were monitored weekly by the bioluminescence IVIS Imaging System 200 using a cryogenically cooled imaging system coupled to a data acquisition computer running Living Image software. Before imaging, animals were anesthetized in an acrylic chamber with 2.5% isoflurane/air mixture and injected i.p. with 40 mg/mL d-luciferin potassium salt in PBS at a dose of 150 mg/kg body weight. After 10 min of incubation with luciferin, mice were placed in a right lateral decubitus position and a digital grayscale animal image was acquired followed by acquisition and overlay of a pseudocolor image representing the spatial distribution of detected photons emerging from active luciferase within the animal. Signal intensity was quantified as the sum of all detected photons within the region of interest per second. Mice were imaged on days 0, 7, 14, 21, 24, and 31 of treatment. Therapy was continued for 4 weeks and animals were sacrificed 1 week later. Primary tumors in the pancreas were excised and the final tumor volume was measured as V = 2 / 3πr3, where r is the mean of the three dimensions (length, width, and depth). The final tumor volumes were initially subjected to one-way ANOVA and then later compared among groups using unpaired Student's t test. Half of the tumor tissue was formalin fixed and paraffin embedded for immunohistochemistry and routine H&E staining. The other half was snap frozen in liquid nitrogen and stored at −80°C. H&E staining confirmed the presence of tumor(s) in each pancreas.[5] |
|
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Peak plasma concentrations of gemcitabine range from 10 to 40 mg/L following a 30-minute intravenous infusion, and are reached at 15 to 30 minutes. One study showed that steady-state concentrations of gemcitabine showed a linear relationship to dose over the dose range 53 to 1000 mg/m2. Gemcitabine triphosphate, the active metabolite of gemcitabine, can accumulate in circulating peripheral blood mononuclear cells. In one study, the Cmax of gemcitabine triphosphate in peripheral blood mononuclear cells occurred within 30 minutes of the end of the infusion period and increased increased proportionally with gemcitabine doses of up to 350 mg/m2. Gemcitabine mainly undergoes renal excretion. Within a week following administration of a single dose of 1000 mg/m2 infused over 30 minutes, about 92-98% of the dose was recovered in urine where 89% of the recovered dose was excreted as difluorodeoxyuridine (dFdU) and less than 10% as gemcitabine. Monophosphate, diphosphate, or triphosphate metabolites of gemcitabine are not detectable in urine. In a single-dose study, about 1% of the administered dose was recovered in the feces. In patients with various solid tumours, the volume of distribution increased with infusion length. The volume of distribution of gemcitabine was 50 L/m2 following infusions lasting less than 70 minutes. For long infusions, the volume of distribution rose to 370 L/m2. Gemcitabine triphosphate, the active metabolite of gemcitabine, accumulates and retains in solid tumour cells _in vitro_ and _in vivo_. It is not extensively distributed to tissues after short infusions that last less than 70 minutes. It is not known whether gemcitabine crosses the blood-brain barrier, but gemcitabine is widely distributed into tissues, including ascitic fluid. In rats, placental and lacteal transfer occurred rapidly at five to 15 minutes following drug administration. Following intravenous infusions lasting less than 70 minutes, clearance ranged from 41 to 92 L/h/m2 in males and ranged from 31 to 69 L/h/m2 in females. Clearance decreases with age. Females have about 30% lower clearance than male patients. Gemcitabine pharmacokinetics are linear and are described by a 2-compartment model. Population pharmacokinetic analyses of combined single and multiple dose studies showed that the volume of distribution of gemcitabine was significantly influenced by duration of infusion and gender. Clearance was affected by age and gender. Differences in either clearance or volume of distribution based on patient characteristics or the duration of infusion result in changes in half-life and plasma concentrations. Protein binding /of gemcitabine/ is very low, less than 10%. It is not known wether gemcitabine or its metabolites are distributed into breast milk. /Elimination is/ renal. 92 to 98% of a single dose of radiolabeled gemcitabine (1000 mg per square meter of body surface area, given over 30 minutes to five patients) was recovered within 1 week, primarily as the inactive uracil metabolite (approximately 89% of the excreted dose) and secondarily as unchanged gemcitabine (less than 10% of the excreted dose). For more Absorption, Distribution and Excretion (Complete) data for GEMCITABINE (8 total), please visit the HSDB record page. Metabolism / Metabolites Following administration and uptake into cancer cells, gemcitabine is initially phosphorylated by deoxycytidine kinase (dCK), and to a lower extent, the extra-mitochondrial thymidine kinase 2 to form gemcitabine monophosphate (dFdCMP). dFdCMP is subsequently phosphorylated by nucleoside kinases to form active metabolites, gemcitabine diphosphate (dFdCDP) and gemcitabine triphosphate (dFdCTP). Gemcitabine is also deaminated intracellularly and extracellularly by cytidine deaminase to its inactive metabolite 2′,2′-difluorodeoxyuridine or 2´-deoxy-2´,2´-difluorouridine (dFdU). Deamination occurs in the blood, liver, kidneys, and other tissues, and this metabolic pathway accounts for most of drug clearance. Gemcitabine undergoes intracellular metabolism, via nucleoside kinases, to produce two active metabolites (gemcitabine diphosphate and gemcitabine triphosphate) and also undergoes deamination to an active uracil metabolite. ... After intravenous injection, gemcitabine is rapidly converted to the inactive metabolite 2'-deoxy-2',2'-difluorouridine by cytidine deaminase. ... Biological Half-Life Following intravenous infusions lasting less than 70 minutes, the terminal half-life ranged from 0.7 to 1.6 hours. Following infusions ranging from 70 to 285 minutes, the terminal half-life ranged from 4.1 to 10.6 hours. Females tend to have longer half-lives than male patients. Gemcitabine triphosphate, the active metabolite of gemcitabine, can accumulate in circulating peripheral blood mononuclear cells. The terminal half-life of gemcitabine triphosphate, the active metabolite, from mononuclear cells ranges from 1.7 to 19.4 hours. The current study was performed in nonhuman primates to determine the plasma and CSF pharmacokinetics of gemcitabine and its inactive metabolite, difluorodeoxyuridine (dFdU) following iv administration. Gemcitabine, 200 mg/kg, was administered iv over 45 min to four nonhuman primates. Serial plasma and CSF samples were obtained prior to, during, and after completion of the infusion for determination of gemcitabine and dFdU concentrations. ... Plasma elimination was rapid with a mean t1/2 of 8 +/- 4 min (mean +/- SD) for gemcitabine and 83 +/- 8 min for dFdU. Gemcitabine total body clearance (ClTB) was 177 +/- 40 mL/min per kg and the Vdss was 5.5 +/- 1.0 L/kg. The maximum concentrations (Cmax) and areas under the time concentration curves (AUC) for gemcitabine and dFdU in plasma were 194 +/- 64 uM and 63.8 +/- 14.6 uM.hr, and 783 +/- 99 uM and 1725 +/- 186 uM.hr, respectively. The peak CSF concentrations of gemcitabine and dFdU were 2.5 +/- 1.4 uM and 32 +/- 41 uM, respectively. The mean CSF:plasma ratio was 6.7% for gemcitabine and 23.8% for dFdU. There is only modest penetration of gemcitabine into the CSF after iv administration. In this study, the plasma pharmacokinetics (PKs) of gemcitabine and dFdU are further explored after gemcitabine doses of 10, 30, and 60 mg/kg administered by intravenous infusion with a loading dose /to dogs/. Gemcitabine displayed linear PKs, while the kinetics of 2',2'-difluorodeoxyuridine (dFdU) were not dose proportional. The overall clearance, volume of distribution at steady-state, and terminal elimination half-life (t(1/2)) for gemcitabine were 0.421 L/hr.kg, 0.822 L/kg, and 1.49 hr, respectively. Plasma concentrations of dFdU peaked at approximately 2 hr postdosing and had a t(1/2) of 14.9 hr. |
|
| Toxicity/Toxicokinetics |
Hepatotoxicity Elevations in serum aminotransferase levels occur in 30% to 90% of patients receiving cyclic therapy with gemcitabine. The elevations are generally mild-to-moderate, asymptomatic and self-limited, frequently resolving without discontinuation or even interruption of therapy. ALT or AST elevations above 5 times the upper limit of the normal range occur in 1-4% of patients yet rarely lead to symptoms or clinically apparent liver injury. Serum bilirubin and alkaline phosphatase elevations are less common, but also typically transient and mild. Despite wide use, gemcitabine has only rarely been implicated in rare cases of acute liver injury with jaundice, and most published cases have been reported in patients with underlying chronic liver disease or extensive hepatic metastases. The clinical features of hepatotoxicity from gemcitabine have not been well described. Most cases were marked by a progressive cholestasis and hepatic failure developing after several cycles of therapy in patients with preexisting chronic liver disease (hepatitis C, alcoholic liver disease) or significant hepatic metastases or local invasion. As with many antineoplastic agents and regimens, therapy with gemcitabine has also been associated rare cases of with reactivation of hepatitis B in persons with preexisting HBsAg in serum. At least one case of sinusoidal obstruction syndrome (veno-occlusive disease) has been reported with use of gemcitabine in a patient with underlying chronic hepatitis C who received no other antineoplastic agent. Likelihood score: C (probable rare cause of clinically apparent liver injury). Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Most sources consider breastfeeding to be contraindicated during maternal antineoplastic drug therapy. It might be possible to breastfeed safely during intermittent gemcitabine therapy with an appropriate period of breastfeeding abstinence; the manufacturer recommends an abstinence period of at least 1 week after the last dose. Chemotherapy may adversely affect the normal microbiome and chemical makeup of breastmilk. Women who receive chemotherapy during pregnancy are more likely to have difficulty nursing their infant. ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk A telephone follow-up study was conducted on 74 women who received cancer chemotherapy at one center during the second or third trimester of pregnancy to determine if they were successful at breastfeeding postpartum. Only 34% of the women were able to exclusively breastfeed their infants, and 66% of the women reported experiencing breastfeeding difficulties. This was in comparison to a 91% breastfeeding success rate in 22 other mothers diagnosed during pregnancy, but not treated with chemotherapy. Other statistically significant correlations included: 1. mothers with breastfeeding difficulties had an average of 5.5 cycles of chemotherapy compared with 3.8 cycles among mothers who had no difficulties; and 2. mothers with breastfeeding difficulties received their first cycle of chemotherapy on average 3.4 weeks earlier in pregnancy. Of the 9 women who received a fluorouracil-containing regimen, 8 had breastfeeding difficulties. Protein Binding Gemcitabine plasma protein binding is less than 10%. Interactions ... /The authors/ present the first case of a nonlung cancer patient experiencing not only acne-like skin toxicity, but subsequently also severe interstitial lung disease during therapy with gemcitabine and erlotinib. Both therapeutic agents were suspected as a possible cause of this adverse event. An interaction between gemcitabine and erlotinib might have also contributed to the pathogenesis of this pulmonary toxicity. Treatment with high-dose steroids was, however, very effective in our patient and a complete recovery appeared within a few days. Thus, pulmonary side effects should be regarded carefully in pancreatic cancer patients receiving palliative therapy with gemcitabine and erlotinib. /The authors/ investigated the possible pharmacokinetic interactions of gemcitabine and oxaliplatin in patients with advanced solid tumors. Ten patients with advanced stage solid tumors were treated with gemcitabine (1500 mg/sq m) as a 30-min intravenous infusion on days 1 and 8, followed by oxaliplatin (130 mg/sq m) as a 4-hr intravenous infusion, on day 8 every 21 days. Pharmacokinetic data for 24 hr after dosing were obtained for both day 1 (gemcitabine without oxaliplatin coadministration) and day 8 (gemcitabine with oxaliplatin) during the first cycle of treatment. Gemcitabine levels in plasma were quantified using a reverse-phase high-performance liquid chromatography assay with ultraviolet detection, and total and ultrafiltrated platinum levels by flameless atomic absorption spectrophotometry with deuterium correction. All pharmacokinetic parameters of gemcitabine seemed to be unchanged when coadministered with oxaliplatin (day 8) compared with pharmacokinetic data of gemcitabine given as a single agent (day 1). The mean (maximum) concentration of gemcitabine on days 1 and 8 was 13.57 (+/-7.42) and 10.23 (+/-5.21) mg/L, respectively (P=0.28), and the mean half-life was 0.32 and 0.44 hr, respectively (P=0.40). Similarly, the P-values for AUC0-24 and the observed clearance were 0.61 and 0.30, respectively. Plasma total and free platinum levels were in agreement with other published data. Gemcitabine disposition appeared to be unaffected by oxaliplatin coadministration because no significant changes in pharmacokinetics between day 1 (gemcitabine without oxaliplatin coadministration) and day 8 (gemcitabine with oxaliplatin) were observed. |
|
| References |
[1]. Cancer Res . 1990 Jul 15;50(14):4417-22. [2]. Clin Cancer Res . 2000 May;6(5):1936-48. [3]. Semin Oncol . 1995 Aug;22(4 Suppl 11):72-9. [4]. Clin Cancer Res . 2005 Sep 15;11(18):6713-21. [5]. Cancer Res . 2007 Apr 15;67(8):3853-61. [6]. Mol Cancer . 2022 May 10;21(1):112. |
|
| Additional Infomation |
Therapeutic Uses Antineoplastic Gemcitabine in combination with paclitaxel is indicated for the first-line treatment of patients with metastatic breast cancer after the failure of prior anthracycline-containing adjuvant chemotherapy, unless anthracyclines are clinically contraindicated. /Included in US product label/ Gemcitabine is indicated as first-line therapy for locally advanced (nonresectable stage II or III) or metastatic (stage IV) adenocarcinoma of the pancreas. It is also indicated as second-line therapy for patients who have previously been treated with fluorouracil. Treatment with gemcitabine is primarily palliative. /Included in US product label/ Gemcitabine is indicated in combination with cisplatin as a first-line therapy for inoperable, locally advanced (Stage IIIA or IIIB) or metastatic (Stage IV) non-small cell lung carcinoma. /Included in US product label/ For more Therapeutic Uses (Complete) data for GEMCITABINE (9 total), please visit the HSDB record page. Drug Warnings A complete blood cell count (CBC), including differential and platelets, should be performed prior to each dose of gemcitabine. If myelosuppression is detected, therapy should be modified or temporarily withheld according to the degree of hematologic toxicity. For patients with absolute granulocyte counts of at least 1000/cu m and platelet counts of at least 100,000/cu m, no adjustment in dosage is necessary. For those with absolute granulocyte counts of 500-999/cu m or platelet counts of 50,000-99,000/cu m, 75% of the full dose should be given weekly. If the absolute granulocyte count is less than 500/cu m or the platelet count is less than 50,000/cu m, the weekly dose should be withheld until the counts exceed these levels. The diagnosis of hemolytic-uremic syndrome should be considered and gemcitabine should be discontinued immediately in patients who develop anemia with evidence of microangiopathic hemolysis, elevation of serum bilirubin or LDH, reticulocytosis, and/or severe thrombocytopenia with or without evidence of renal failure (e.g., elevation of serum creatinine or BUN). Gemcitabine should be discontinued immediately and appropriate supportive care (e.g., diuretics, corticosteroids) provided promptly in patients who develop severe adverse pulmonary effects. The bone marrow depressant effects of gemcitabine may result in an increased incidence of microbial infection, delayed healing, and gingival bleeding. Dental work, whenever possible, should be completed prior to the initiation of therapy or deferred until blood counts have returned to normal. Patients should be instructed in proper hygiene during treatment, including caution in use of regular toothbrushes, dental floss, and toothpicks. FDA Pregnancy Risk Category: D /POSITIVE EVIDENCE OF RISK. Studies in humans, or investigational or post-marketing data, have demonstrated fetal risk. Nevertheless, potential benefits from the use of the drug may outweigh the potential risk. For example, the drug may be acceptable if needed in a life-threatening situation or serious disease for which safer drugs cannot be used or are ineffective./ Pharmacodynamics Gemcitabine is a nucleoside analog that mediates its antitumour effects by promoting apoptosis of malignant cells undergoing DNA synthesis. More specifically, it blocks the progression of cells through the G1/S-phase boundary. Gemcitabine demonstrated cytotoxic effects against a broad range of cancer cell lines _in vitro_. It displayed schedule-dependent antitumour activity in various animal models and xenografts from human non-small cell lung cancer (NSCLC) and pancreatic cancer. Therefore, the antineoplastic effects of gemcitabine are enhanced through prolonged infusion time rather than higher dosage. Gemcitabine inhibited the growth of human xenografts from carcinoma of the lung, pancreas, ovaries, head and neck, and breast. In mice, gemcitabine inhibited the growth of human tumour xenografts from the breast, colon, lung or pancreas by 69 to 99%. In clinical trials of advanced NSCLC, gemcitabine monotherapy produced objective response rates ranging from 18 to 26%, with a median duration of response ranging from 3.3 to 12.7 months. Overall median survival time was 6.2 to 12.3 months. The combined use of cisplatin and gemcitabine produced better objective response rates compared to monotherapy. In patients with advanced pancreatic cancer, objective response rates in patients ranged from 5.to 12%, with a median survival duration of 3.9 to 6.3 months. In Phase II trials involving patients with metastatic breast cancer, treatment with gemcitabine alone or with adjuvant chemotherapies resulted in response rate ranging from 13 to 42% and median survival duration ranging from 11.5 to 17.8 months. In metastatic bladder cancer, gemcitabine has a response rate 20 to 28%. In Phase II trials of advanced ovarian cancer, patients treated with gemcitabine had response rate of 57.1%, with progression free survival of 13.4 months and median survival of 24 months. Gemcitabine causes dose-limiting myelosuppression, such as anemia, leukopenia, neutropenia, and thrombocytopenia; however, events leading to discontinuation tend to occur less than 1% of the patients. Gemcitabine can elevate ALT, AST and alkaline phosphatase levels. |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.62 mg/mL (9.95 mM) (saturation unknown) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.62 mg/mL (9.95 mM) (saturation unknown) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.58 mg/mL (9.80 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: ≥ 2.08 mg/mL (7.90 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL of PEG300 and mix evenly; then add 50 μL of Tween-80 to the above solution and mix evenly; then add 450 μL of normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 5: ≥ 2.08 mg/mL (7.90 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 6: ≥ 2.08 mg/mL (7.90 mM) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 7: ≥ 2.62 mg/mL (9.95 mM) (saturation unknown) in 5% DMSO + 40% PEG300 + 5% Tween80 + 50% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 8: 20 mg/mL (75.99 mM) in 0.5%HPMC + 1%Tween80 (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.7994 mL | 18.9970 mL | 37.9939 mL | |
| 5 mM | 0.7599 mL | 3.7994 mL | 7.5988 mL | |
| 10 mM | 0.3799 mL | 1.8997 mL | 3.7994 mL |