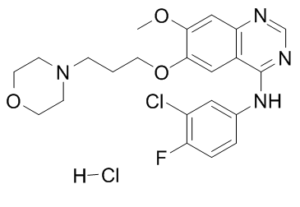
Gefitinib hydrochloride, the HCl salt of Gefitinib (formerly also known as ZD1839 or trade name: Iressa), is a potent and orally-bioavailable EGFR inhibitor for Tyr1173, Tyr992, Tyr1173 and Tyr992 in the NR6wtEGFR and NR6W cells with IC50 of 37 nM, 37nM, 26 nM and 57 nM, respectively. Gefitinib exhibits anti-angiogenic activities in a wide range of human tumor types, including head and neck, prostate, breast, ovarian, colon, small-cell lung and non-small-cell lung cancer. In May 2003, the FDA approved Gefitinib for non-small cell lung cancer (NSCLC). It was approved as monotherapy for the treatment of patients with locally advanced or metastatic NSCLC after failure of both platinum-based and docetaxel chemotherapies. i.e. as a third-line therapy.
Physicochemical Properties
| Molecular Formula | C22H24N4O3FCL.HCL |
| Molecular Weight | 483.3633 |
| Exact Mass | 482.128 |
| CAS # | 184475-55-6 |
| Related CAS # | Gefitinib;184475-35-2 |
| PubChem CID | 19077490 |
| Appearance | Typically exists as white to off-white solids at room temperature |
| Boiling Point | 607.7ºC at 760 mmHg |
| Flash Point | 321.3ºC |
| Vapour Pressure | 4.9E-15mmHg at 25°C |
| LogP | 5.088 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 8 |
| Rotatable Bond Count | 8 |
| Heavy Atom Count | 32 |
| Complexity | 545 |
| Defined Atom Stereocenter Count | 0 |
| SMILES | ClC1=C(C=CC(NC2=NC=NC3=C2C=C(C(OC)=C3)OCCCN4CCOCC4)=C1)F.[H]Cl |
| InChi Key | QUINXWLATMJDQF-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C22H24ClFN4O3.ClH/c1-29-20-13-19-16(12-21(20)31-8-2-5-28-6-9-30-10-7-28)22(26-14-25-19)27-15-3-4-18(24)17(23)11-15;/h3-4,11-14H,2,5-10H2,1H3,(H,25,26,27);1H |
| Chemical Name | N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine;hydrochloride |
| Synonyms | ZD1839 HCl; ZD 1839 HCl; ZD-1839 HCl; Gefitinib HCl; trade name: Iressa.Gefitinib hydrochloride; 184475-55-6; gefitinib hcl; Gefitinib (hydrochloride); ZD-1839 hydrochloride; Gefitinib hydrochloride salt; N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3-morpholin-4-ylpropoxy)quinazolin-4-amine;hydrochloride; 4-Quinazolinamine,N-(3-chloro-4-fluorophenyl)-7-methoxy-6-[3-(4-morpholinyl)propoxy]-,monohydrochloride; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | EGFR tyrosine kinase | ||
| ln Vitro |
After long-term exposure of EGFRvIII-expressing cells, gefitinib (0.01-0.1 mM) increases the phosphotyrosine load of the receptor, increases signaling to ERK, and stimulates proliferation and anchorage-independent growth. This effect is likely caused by the induction of EGFRvIII dimerization. Conversely, EGFRvIII phosphotyrosine load, EGFRvIII-mediated proliferation, and anchorage-independent growth are all markedly reduced by gefitinib (1-2 mM)[1]. With an IC50 of 20 nM, gefitinib (ZD1839) prevents these EGF-driven untransformed cells from growing monolayer[2]. With an IC50 of 2 μM, gefitinib inhibits the growth of GLC82 and CALU-3 cells[3].
Epidermal growth factor receptor (EGFR) is frequently amplified and/or mutated in a number of human tumours and abnormal signalling from this receptor is believed to contribute to the malignant phenotype seen in these tumours. Gefitinib is a small molecule inhibitor that specifically binds and inhibits the EGFR tyrosine kinase and has been shown to inhibit the growth, proliferation, survival and invasion of a range of tumour cells overexpressing EGFR. However, clinical response to gefitinib has failed to correlate with EGFR levels and activity, indicating that other molecular mechanisms such as downstream signalling and mutations could be of importance in predicting clinical response. We therefore investigated the effect of the specific EGFR inhibitor gefitinib on the phosphorylation level, signalling and growth of cells expressing the naturally occurring constitutively active EGFR variant EGFRvIII, a low nontransforming level of EGFR and a high transforming level of EGFR. Results show that levels of gefitinib sufficient to suppress EGFR phosphorylations, EGFR-mediated proliferation and EGFR-mediated anchorage-independent growth are not sufficient to inhibit these features in cells expressing EGFRvIII. Furthermore, the data indicate that long-term exposure of EGFRvIII-expressing cells to low concentrations of gefitinib (0.01-0.1 microM) result in increased phosphotyrosine load of the receptor, increased signalling to ERK and stimulation of proliferation and anchorage-independent growth, presumably by inducing EGFRvIII dimerisation. Higher concentrations of gefitinib (1-2 microM), on the other hand, significantly decreased EGFRvIII phosphotyrosine load, EGFRvIII-mediated proliferation and anchorage-independent growth. Further studies are needed to investigate the implications of these important findings in the clinical setting. [2] The epidermal growth factor receptor (EGFR) is commonly overexpressed in many human tumors and provides a new target for anticancer drug development. ZD1839 ("Iressa"), a quinazoline tyrosine kinase inhibitor selective for the EGFR, has shown good activity in preclinical studies and in the early phase of clinical trials. However, because it remains unclear which tumor types are the best targets for treatment with this agent, the molecular characteristics that correlate with tumor sensitivity to ZD1839 have been studied. In a panel of human breast cancer and other epithelial tumor cell lines, HER2-overexpressing tumors were particularly sensitive to ZD1839. Growth inhibition of these tumor cell lines was associated with the dephosphorylation of EGFR, HER2, and HER3, accompanied by the loss of association of HER3 with phosphatidylinositol 3-kinase, and down-regulation of Akt activity. These studies suggest that HER2-overexpressing tumors are particularly susceptible to the inhibition of HER family tyrosine kinase signaling and suggest novel strategies to treat these particularly aggressive tumors.[3] |
||
| ln Vivo |
When metformin and gefitinib (150 mg/kg, po) are given to nude mice containing H1299 or CALU-3 GEF-R cells that are cultured subcutaneously as tumor xenografts, the tumor growth is significantly reduced[3]. While gefitinib therapy attenuates fibrotic lung remodeling due to the reduction of lung fibroblast proliferation, it increases lung inflammation in irradiated rats, including inflammatory cell infiltration and pro-inflammatory cytokine expression[4]. In studies with mice bearing a range of human tumor-derived xenografts, ZD1839 given p.o. once a day inhibited tumor growth in a dose-dependent manner. The level of expression of EGFR did not determine xenograft tumor sensitivity to ZD1839. Long-term ZD1839 (>3 months) treatment of mice bearing A431 xenografts was well tolerated, and ZD1839 completely inhibited tumor growth and induced regression of established tumors. No drug-resistant tumors appeared during ZD1839 treatment, but some tumors regrew after drug withdrawal. These studies indicate the potential utility of ZD1839 in the treatment of many human tumors and indicate that continuous once-a-day p.o. dosing might be a suitable therapeutic regimen.[1] Gefitinib treatment increased the infiltration of inflammatory cells, which produced more pro-inflammatory cytokines (IL-6, IL-1β), in the lungs of the irradiated rats on days 15 and 57, while gefitinib treatment reduced collagen content of the lungs in irradiated rats and decreased proliferation and EGFR expression in the lung fibroblasts from irradiated rats on day 57. Conclusions: In irradiated rats, gefitinib treatment augmented lung inflammation, including inflammatory cell infiltration and pro-inflammatory cytokine expression, while gefitinib treatment attenuated fibrotic lung remodeling due to the inhibition of lung fibroblast proliferation [5]. |
||
| Cell Assay |
Purpose: EGF receptor (EGFR) tyrosine kinase inhibitors (TKI) have been found to be effective against lung cancer, but clinical resistance to these agents has developed as their usage has increased. Metformin is a widely used antidiabetic drug and also displays significant growth-inhibitory and proapoptotic effects in several cancer models, alone or in combination with chemotherapeutic drugs. Experimental design: The effects of gefitinib, a selective EGFR-TKI, and metformin on a panel of non-small cell lung cancer (NSCLC) cell lines were assessed by using MTT, bromide assay, flow cytometry, anchorage-independent growth, coimmunoprecipitation, and Western blot analysis. Results: The combination of metformin with gefitinib induced a strong antiproliferative and proapoptotic effect in NSCLC cell lines that harbored wild-type LKB1 gene. Treatment with metformin as single agent, however, induced an activation and phosphorylation of mitogen-activated protein kinase (MAPK) through an increased C-RAF/B-RAF heterodimerization. The inhibition of EGFR phosphorylation and of downstream signaling by adding gefitinib to metformin treatment abrogated this phenomenon and induced a strong apoptotic effect in vitro and in vivo. Conclusions: Metformin and gefitinib are synergistic in LKB1 wild-type NSCLC cells. However, further studies are required to investigate better the effect of metformin action on the RAS/RAF/MAPK pathway and the best context in which to use metformin in combination with molecular targeted agents.[4] |
||
| Animal Protocol |
|
||
| References |
[1]. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002 Oct 15;62(20):5749-54. [2]. Differential response to gefitinib of cells expressing normal EGFR and the mutant EGFRvIII. Br J Cancer. 2005 Oct 17;93(8):915-23. [3]. The tyrosine kinase inhibitor ZD1839 ("Iressa") inhibits HER2-driven signaling and suppresses the growth of HER2-overexpressing tumor cells. Cancer Res. 2001 Oct 1;61(19):7184-8. [4]. Synergistic effects of metformin treatment in combination with gefitinib, a selective EGFR tyrosine kinase inhibitor, in LKB1 wild-type NSCLC cell lines. Clin Cancer Res. 2013 Jul 1;19(13):3508-19. [5]. Epidermal growth factor receptor-tyrosine kinase inhibitor (gefitinib) augments pneumonitis, but attenuates lung fibrosis in response to radiation injury in rats. J Med Invest. 2012;59(1-2):174-85. |
||
| Additional Infomation | The epidermal growth factor receptor (EGFR) is a promising target for anticancer therapy because of its role in tumor growth, metastasis and angiogenesis, and tumor resistance to chemotherapy and radiotherapy. We have developed a low-molecular-weight EGFR tyrosine kinase inhibitor (EGFR-TKI), ZD1839 (Iressa(2) ). ZD1839, a substituted anilinoquinazoline, is a potent EGFR-TKI (IC(50) = 0.033 micro M) that selectively inhibits EGF-stimulated tumor cell growth (IC(50) = 0.054 micro M) and that blocks EGF-stimulated EGFR autophosphorylation in tumor cells. In studies with mice bearing a range of human tumor-derived xenografts, ZD1839 given p.o. once a day inhibited tumor growth in a dose-dependent manner. The level of expression of EGFR did not determine xenograft tumor sensitivity to ZD1839. Long-term ZD1839 (>3 months) treatment of mice bearing A431 xenografts was well tolerated, and ZD1839 completely inhibited tumor growth and induced regression of established tumors. No drug-resistant tumors appeared during ZD1839 treatment, but some tumors regrew after drug withdrawal. These studies indicate the potential utility of ZD1839 in the treatment of many human tumors and indicate that continuous once-a-day p.o. dosing might be a suitable therapeutic regimen. [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
|
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.0689 mL | 10.3443 mL | 20.6885 mL | |
| 5 mM | 0.4138 mL | 2.0689 mL | 4.1377 mL | |
| 10 mM | 0.2069 mL | 1.0344 mL | 2.0689 mL |