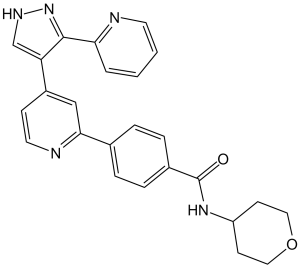
GW788388 (GW-788388; GW 788388) is an inhibitor of ALK5/TGF-β type II receptor with potential antifibrotic activity. It inhibits ALK5/TGF-β type II receptor with an IC50 of 18 nM in a cell-free assay.
Physicochemical Properties
| Molecular Formula | C25H23N5O2 | |
| Molecular Weight | 425.48 | |
| Exact Mass | 425.185 | |
| CAS # | 452342-67-5 | |
| Related CAS # |
|
|
| PubChem CID | 10202642 | |
| Appearance | Off-white to gray solid powder | |
| Density | 1.3±0.1 g/cm3 | |
| Boiling Point | 683.2±55.0 °C at 760 mmHg | |
| Flash Point | 367.0±31.5 °C | |
| Vapour Pressure | 0.0±2.1 mmHg at 25°C | |
| Index of Refraction | 1.680 | |
| LogP | 1.88 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 5 | |
| Rotatable Bond Count | 5 | |
| Heavy Atom Count | 32 | |
| Complexity | 605 | |
| Defined Atom Stereocenter Count | 0 | |
| InChi Key | SAGZIBJAQGBRQA-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C25H23N5O2/c31-25(29-20-9-13-32-14-10-20)18-6-4-17(5-7-18)23-15-19(8-12-27-23)21-16-28-30-24(21)22-3-1-2-11-26-22/h1-8,11-12,15-16,20H,9-10,13-14H2,(H,28,30)(H,29,31) | |
| Chemical Name | N-(oxan-4-yl)-4-[4-(5-pyridin-2-yl-1H-pyrazol-4-yl)pyridin-2-yl]benzamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
GW788388 targets TGF-beta type I receptor (ALK5) with an IC50 of 18 nM, and TGF-beta type II receptor (TβRII) with weak inhibitory activity (IC50=1.6 μM) [3] GW788388 exhibits high selectivity for ALK5 over other ALK family members: ALK1 (IC50=1.6 μM), ALK2 (IC50=2.4 μM), ALK3 (IC50=5.3 μM), ALK4 (IC50>10 μM), ALK6 (IC50>10 μM), ALK7 (IC50>10 μM) [3] |
| ln Vitro |
In vitro activity: GW788388 shows anti-TGF-β activity with IC50 of 93 nM in cellular assay. GW788388 shows some inhibitory to activin type II receptor (ActRII) but no inhibitory to bone morphogenic protein (BMP) type II receptor. GW788388 shows no toxicity in Namru murine mammary gland (NMuMG), MDA-MB-231, renal cell carcinoma (RCC)4, and U2OS cells at 4 nM to 15 μM. GW788388 blocks TGF-β-induced Smad activation and target gene expression, while decreasing epithelial-mesenchymal transitions and fibrogenesis. GW788388 inhibits ALK5, ALK4, ALK7 and TGF-β-mediated growth arrest. Kinase Assay: GW788388 binding to ALK5 is tested on purified recombinant GST−ALK5 (residues 198-503). Displacement of rhodamine green fluorescently labeled ATP competitive inhibitor by different concentrations of GW788388 is used to calculate a binding pIC50. GST−ALK5 is added to a buffer containing 62.5 mM N-(2-hydroxyethyl)piperazine-N-2-ethanesulfonic acid (Hepes), pH 7.5, 1 mM dithiothreitol (DTT), 12.5 mM MgCl2, 1.25 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid (CHAPS), and 1 nM rhodamine green-labeled ligand so that the final ALK5 concentration is 10 nM based on active-site titration of the enzyme. The enzyme/ligand reagent (40 μL) is added to 384-well assay plates containing 1 μL of different concentrations of GW788388. The plates are read immediately on a LJL Acquest fluorescence reader with excitation, emission, and dichroic filters of 485, 530, and 505 nm, respectively. The fluorescence polarization for each well is calculated by the Acquest and is then imported into curve-fitting software for construction of concentration−response curves. Cell Assay: Cell viability/proliferation assays (Namru murine mammary gland (NMuMG), MDA-MB-231, renal cell carcinoma (RCC)4, and U2OS cells) are done according to the manufactures instructions (CellTiter 96 Aqueous One Solution Cell Proliferation Assay). Viability and proliferation are measured after 72 hours GW788388 treatment in the presence or absence of TGF-β. GW788388 dose-dependently inhibited ALK5 kinase activity, achieving 50% inhibition at 18 nM [3] GW788388 blocked TGF-β1-induced Smad2/3 phosphorylation in Mv1Lu cells with an EC50 of 41 nM, and suppressed TGF-β-responsive luciferase reporter activity (3TP-Lux) by 90% at 1 μM [3] GW788388 (1-10 μM) reduced TGF-β1-induced α-SMA and collagen type I mRNA expression in rat renal fibroblasts (NRK-49F) by 45-78% [1] GW788388 (0.1-10 μM) inhibited TGF-β1-induced proliferation of cardiac fibroblasts (CFs) with an IC50 of 0.8 μM, and reduced collagen type I and III protein levels by 35-62% [2] GW788388 (1 μM) blocked TGF-β1-mediated epithelial-mesenchymal transition (EMT) in HK-2 renal tubular epithelial cells, as evidenced by preserved E-cadherin expression and reduced vimentin levels [1] |
| ln Vivo |
For five weeks, db/db mice receiving oral GW788388 treatment had a significant reduction in renal fibrosis and mRNA levels of important mediators of renal extracellular matrix deposition [1]. In rats with myocardial infarction, GW788388 (50 mg/kg/day, p.o.) significantly reduced contractile dysfunction while also attenuating activated (phosphorylated) Smad2 (P < 0.01), α-smooth muscle actin (P < 0.001), and Collagen I (P < 0.05) [2]. Taken twice daily (bid), GW788388 lowers collagen IA1 expression by 80%. The dosage is 1 mg/kg. In a puromycin aminonucleoside-induced renal fibrosis model, GW788388, when given orally at 10 mg/kg once daily (uid), significantly decreased the expression of collagen IA1 mRNA [3]. GW788388 (1, 3, 10 mg/kg, oral, daily for 14 days) reduced renal fibrosis in UUO (unilateral ureteral obstruction) rats: renal collagen content decreased by 28-65%, α-SMA-positive cells reduced by 32-68%, and Col1a1 mRNA expression downregulated by 40-72% [1] GW788388 (10 mg/kg, oral) improved renal function in UUO rats: serum creatinine reduced by 35% and blood urea nitrogen (BUN) decreased by 38% compared to the vehicle group [1] GW788388 (3, 10 mg/kg, oral, daily for 4 weeks) attenuated cardiac dysfunction in myocardial infarction (MI) mice: left ventricular ejection fraction (LVEF) increased by 18-32%, left ventricular fractional shortening (LVFS) improved by 20-35%, and cardiac fibrosis area reduced by 30-58% [2] GW788388 (10 mg/kg, oral) reduced myocardial TGF-β1 levels (42% decrease) and Smad2/3 phosphorylation (55% decrease) in MI mice [2] |
| Enzyme Assay |
Recombinant ALK5 kinase was incubated with a specific peptide substrate, ATP, and serial concentrations of GW788388 (0.001-10 μM) in kinase assay buffer at 30°C for 60 minutes. Phosphorylated substrate was detected using a radioactive scintillation assay, and IC50 was calculated by fitting the dose-response inhibition curve [3] Recombinant TβRII and other ALK family kinases (ALK1-7) were incubated with their respective peptide substrates, ATP, and GW788388 (0.001-100 μM) under the same conditions as ALK5. Kinase activity inhibition was quantified, and IC50 values were determined to evaluate selectivity [3] |
| Cell Assay |
Mv1Lu cells transfected with 3TP-Lux reporter plasmid and Renilla luciferase plasmid were seeded in 96-well plates. After 24 hours, cells were treated with GW788388 (0.01-10 μM) and TGF-β1 (5 ng/mL) for 18 hours. Luciferase activity was measured using a dual-luciferase assay system, with results normalized to Renilla luciferase [3] NRK-49F renal fibroblasts were seeded in 6-well plates and serum-starved for 12 hours. Cells were pre-treated with GW788388 (1-10 μM) for 1 hour, then stimulated with TGF-β1 (5 ng/mL) for 24 hours. Total RNA was extracted for qPCR analysis of α-SMA and Col1a1 mRNA expression (GAPDH as reference) [1] Cardiac fibroblasts (CFs) were isolated from neonatal mice and seeded in 96-well plates (5×10^3 cells/well). Cells were treated with GW788388 (0.1-10 μM) and TGF-β1 (5 ng/mL) for 48 hours. Cell proliferation was assessed using a cell counting kit, and collagen protein levels were detected by western blot [2] HK-2 cells were seeded in 6-well plates and pre-treated with GW788388 (1 μM) for 1 hour, followed by TGF-β1 (10 ng/mL) stimulation for 48 hours. E-cadherin and vimentin protein levels were analyzed by western blot [1] |
| Animal Protocol |
Dissolved in 4% DMSO and 96% [0.5% HPMC/5% Tween/20%HCl (1 M) in NaH2PO4 (0.1 M); 3 or 10 mg/kg; oral administration Sprague-Dawley rats with dimethylnitrosamine- (DMN-) induced liver disease or puromycin aminonucleoside-induced renal fibrosis UUO-induced renal fibrosis model: Male Sprague-Dawley (SD) rats (200-250 g) underwent unilateral ureteral ligation. One day after surgery, GW788388 was dissolved in 0.5% methylcellulose and administered orally at doses of 1, 3, or 10 mg/kg once daily for 14 days. The vehicle group received 0.5% methylcellulose alone. Rats were euthanized, and kidneys were collected for histological and molecular analysis [1] MI-induced cardiac dysfunction model: Male C57BL/6 mice (8-10 weeks old) underwent left anterior descending coronary artery ligation to induce MI. Three days after MI, GW788388 was dissolved in 0.5% methylcellulose and given orally at 3 or 10 mg/kg once daily for 4 weeks. Cardiac function was evaluated by echocardiography before sacrifice, and hearts were harvested for fibrosis and molecular analysis [2] Pharmacokinetic study: Male SD rats (250-300 g) were randomly divided into oral (10 mg/kg) and intravenous (2 mg/kg) groups. GW788388 was dissolved in DMSO/PEG400/normal saline (10:40:50, v/v/v) for intravenous administration, and in 0.5% methylcellulose for oral administration. Blood samples were collected at 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours post-administration, and plasma drug concentrations were measured by LC-MS/MS [3] |
| ADME/Pharmacokinetics |
GW788388 showed oral bioavailability of 30% in SD rats [3] GW788388 had a terminal half-life (t1/2) of 2.3 hours (intravenous, 2 mg/kg) and 3.1 hours (oral, 10 mg/kg) in rats [3] GW788388 reached a maximum plasma concentration (Cmax) of 152 ng/mL at 1.2 hours (Tmax) after oral administration of 10 mg/kg in rats [3] The area under the plasma concentration-time curve (AUC0-24h) was 320 ng·h/mL (intravenous, 2 mg/kg) and 890 ng·h/mL (oral, 10 mg/kg) in rats [3] GW788388 had a volume of distribution (Vd) of 1.8 L/kg and total body clearance (CL) of 4.5 mL/min/kg in rats (intravenous administration) [3] |
| Toxicity/Toxicokinetics |
GW788388 had a plasma protein binding rate of >99% in rat and human plasma [3] In rats and mice treated with GW788388 (up to 10 mg/kg, oral, for 4 weeks), no significant weight loss or abnormal clinical signs were observed [1,2,3] Serum levels of ALT, AST, BUN, and Cr in GW788388-treated animals were within the normal range, with no significant difference from the vehicle group [1,2] |
| References |
[1]. Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int, 2008, 73(6), 705-715. [2]. Targeted inhibition of activin receptor-like kinase 5 signaling attenuates cardiac dysfunction following myocardial infarction. Am J Physiol Heart Circ Physiol, 2010, 298(5), H1415-1425. [3]. Discovery of 4-{4-[3-(pyridin-2-yl)-1H-pyrazol-4-yl]pyridin-2-yl}-N-(tetrahydro-2H- pyran-4-yl)benzamide (GW788388): a potent, selective, and orally active transforming growth factor-beta type I receptor inhibitor. J Med Chem. 2006, 49. |
| Additional Infomation |
GW788388 is a synthetic small molecule with the chemical structure 4-{4-[3-(pyridin-2-yl)-1H-pyrazol-4-yl]pyridin-2-yl}-N-(tetrahydro-2H-pyran-4-yl)benzamide [3] GW788388 exerts its inhibitory effect by binding to the ATP-binding pocket of ALK5, preventing ATP binding and subsequent kinase activation, thereby blocking the TGF-β/Smad signaling pathway [3] GW788388 is orally active, which makes it a potential therapeutic agent for fibrotic diseases and cardiac dysfunction associated with excessive TGF-β signaling [1,2,3] GW788388 attenuates renal fibrosis by inhibiting the activation of renal fibroblasts and EMT of tubular epithelial cells [1] GW788388 improves post-MI cardiac function by reducing cardiac fibrosis and suppressing TGF-β-mediated Smad signaling in the myocardium [2] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.88 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.88 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.88 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 30% PEG400+0.5% Tween80+5% Propylene glycol: 30 mg/mL (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.3503 mL | 11.7514 mL | 23.5029 mL | |
| 5 mM | 0.4701 mL | 2.3503 mL | 4.7006 mL | |
| 10 mM | 0.2350 mL | 1.1751 mL | 2.3503 mL |