GSK864 is a novel, potent and selective isocitrate dehydrogenase 1 (IDH1) mutant inhibitor; inhibits IDH1 mutants R132C, R132H, and R132G with IC50 values of 8.8, 15.2 and 16.6 nM. Neomorphic mutations in isocitrate dehydrogenase 1 (IDH1) are driver mutations in acute myeloid leukemia (AML) and other cancers. GSK864 has been shown to be selective in vitro for IDH1 over other classes of proteins (7TMs, ion channels, kinases) and chemoproteomic studies with GSK321, an analog of GSK864, confirm selective binding of IDH1 by this chemical series. GSK864 has a pharmacokinetic profile suitable for in vivo studies.
Physicochemical Properties
| Molecular Formula | C30H31FN6O4 |
| Molecular Weight | 558.603349924088 |
| Exact Mass | 558.239 |
| CAS # | 1816331-66-4 |
| PubChem CID | 91864701 |
| Appearance | White to off-white solid powder |
| Density | 1.4±0.1 g/cm3 |
| Boiling Point | 782.9±60.0 °C at 760 mmHg |
| Flash Point | 427.3±32.9 °C |
| Vapour Pressure | 0.0±2.7 mmHg at 25°C |
| Index of Refraction | 1.661 |
| LogP | 0.01 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 41 |
| Complexity | 967 |
| Defined Atom Stereocenter Count | 1 |
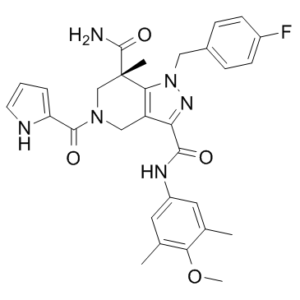
| SMILES | CC1=CC(=CC(=C1OC)C)NC(=O)C2=NN(C3=C2CN(C[C@]3(C)C(=O)N)C(=O)C4=CC=CN4)CC5=CC=C(C=C5)F |
| InChi Key | DUCNNEYLFOQFSW-PMERELPUSA-N |
| InChi Code | InChI=1S/C30H31FN6O4/c1-17-12-21(13-18(2)25(17)41-4)34-27(38)24-22-15-36(28(39)23-6-5-11-33-23)16-30(3,29(32)40)26(22)37(35-24)14-19-7-9-20(31)10-8-19/h5-13,33H,14-16H2,1-4H3,(H2,32,40)(H,34,38)/t30-/m0/s1 |
| Chemical Name | (S)-1-(4-fluorobenzyl)-N3-(4-methoxy-3,5-dimethylphenyl)-7-methyl-5-(1H-pyrrole-2-carbonyl)-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine-3,7-dicarboxamide |
| Synonyms | GSK864; GSK-864; GSK 864. |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Mutant isocitrate dehydrogenase 1 (IDH1) (IDH1 R132H: IC50 = 0.03 μM; IDH1 R132C: IC50 = 0.05 μM; IDH1 R132G: IC50 = 0.07 μM; Wild-type IDH1: IC50 > 10 μM) [1] |
| ln Vitro |
GSK864 had an EC50 of 320 nM and reduced the formation of 2-HG in R132C IDH1 mutant HT1080 fibrosarcoma cells by LCMS/MS analysis [1]. 1. Selective inhibition of mutant IDH1 enzyme activity: GSK864 is a potent and selective inhibitor of oncogenic IDH1 mutants. It inhibits recombinant IDH1 R132H (the most common AML-associated IDH1 mutation) with an IC50 of 0.03 μM, IDH1 R132C with IC50 = 0.05 μM, and IDH1 R132G with IC50 = 0.07 μM. The compound shows minimal activity against wild-type IDH1 (IC50 > 10 μM) and no significant inhibition of IDH2 (wild-type or mutant R140Q/R172K) at concentrations up to 10 μM, demonstrating high target selectivity [1] 2. Suppression of 2-hydroxyglutarate (2-HG) production: In IDH1 R132H-positive AML cell lines (e.g., HT1080, SW1353) and primary AML patient blasts, GSK864 dose-dependently reduces oncogenic 2-HG levels. In HT1080 cells, the EC50 for 2-HG inhibition is 0.12 μM; at 1 μM, 2-HG production is reduced by >90% compared to vehicle control. No significant effect on 2-HG levels is observed in wild-type IDH1-expressing cells (e.g., U937) [1] 3. Induction of leukemia cell differentiation: GSK864 (0.1–1 μM) induces differentiation of IDH1 R132H-positive AML cells (HT1080, primary patient blasts) into mature myeloid cells. Flow cytometry analysis shows increased expression of myeloid differentiation markers CD11b and CD14: at 0.5 μM, CD11b-positive cells increase from 15 ± 3% to 78 ± 5% in HT1080 cells. Morphological analysis confirms the presence of mature granulocyte-like features (segmented nuclei, cytoplasmic granulation) [1] 4. Inhibition of AML cell proliferation and induction of cell cycle arrest: In IDH1 mutant AML cell lines (HT1080, SW1353), GSK864 inhibits cell proliferation with EC50 values of 0.15 μM and 0.21 μM, respectively. Cell cycle analysis by flow cytometry shows G0/G1 phase arrest (45 ± 4% vs. 28 ± 3% in vehicle control) and reduced S phase fraction (30 ± 3% vs. 45 ± 4%) at 1 μM. No significant antiproliferative effect is observed in wild-type IDH1 AML cell lines (U937, HL-60) at concentrations up to 10 μM [1] 5. Apoptosis induction in IDH1 mutant AML cells: GSK864 (1 μM) induces apoptosis in HT1080 cells, with apoptotic cells increasing from 5 ± 2% (vehicle) to 35 ± 4% (Annexin V/PI staining) after 72 hours. Western blot analysis shows upregulation of cleaved caspase-3 and PARP, confirming caspase-dependent apoptosis [1] |
| ln Vivo |
Mice peripheral blood samples were able to sustain significant quantities of GSK864 for a whole day following the intraperitoneal (IP) treatment of CD-1 mice. R132C or R132H IDH1 mutant transplanted animals treated with GSK864 were projected to have more huCD45+ CD38+ cells in their bone marrow (BM cells) according to analysis of early secretory marker expression [1]. 1. Efficacy in IDH1 R132H-positive AML xenograft model: NOD-SCID mice were subcutaneously implanted with HT1080 cells (IDH1 R132H-positive). When tumors reached 100–150 mm³, mice were randomized into vehicle control and GSK864 treatment groups (10 mg/kg, 30 mg/kg, oral gavage, once daily for 21 days). The 30 mg/kg dose significantly inhibited tumor growth, with a tumor growth inhibition (TGI) rate of 78 ± 6% on day 21. Tumor weights at study end were 0.32 ± 0.08 g (30 mg/kg) vs. 1.45 ± 0.21 g (vehicle). 2-HG levels in tumor tissue were reduced by 85 ± 7% at 30 mg/kg [1] 2. Survival extension in systemic AML xenograft model: NOD-SCID mice were intravenously injected with primary IDH1 R132H-positive AML blasts from patients. Mice were treated with GSK864 (30 mg/kg, oral, daily) starting 7 days post-inoculation. Median survival was extended from 32 ± 3 days (vehicle) to 58 ± 5 days (treatment group), representing a 81% increase in survival time. Flow cytometry of bone marrow and peripheral blood showed reduced leukemia cell burden (25 ± 4% vs. 78 ± 6% in vehicle) [1] 3. Pharmacodynamic effect in vivo: In the subcutaneous xenograft model, GSK864 (30 mg/kg) treatment resulted in increased CD11b expression in tumor cells (65 ± 5% vs. 20 ± 3% in vehicle), confirming induction of differentiation in vivo. Histopathological analysis of tumors showed increased mature myeloid cells and reduced mitotic index [1] |
| Enzyme Assay |
1. Recombinant IDH1 mutant enzyme activity assay: - Recombinant human IDH1 mutants (R132H, R132C, R132G) and wild-type IDH1 were purified and resuspended in assay buffer containing Tris-HCl, MgCl₂, and NADP⁺ (cofactor for IDH1 activity). - Serial concentrations of GSK864 (0.001–10 μM) were pre-incubated with the enzyme (100 nM) for 20 minutes at 37°C. - The reaction was initiated by adding D-isocitrate (substrate, 1 mM), and the mixture was incubated at 37°C for 60 minutes. - The production of NADPH (byproduct of IDH1-catalyzed reaction) was measured by fluorescence spectroscopy (excitation 340 nm, emission 460 nm) to quantify enzyme activity. - The percentage of enzyme activity (relative to vehicle control) was plotted against the log concentration of GSK864, and IC50 values were calculated from the dose-response curves [1] 2. 2-HG production inhibition assay in recombinant enzyme system: - Recombinant IDH1 R132H enzyme was incubated with D-isocitrate and NADP⁺ in the presence of GSK864 (0.005–5 μM) at 37°C for 2 hours. - The reaction was terminated by adding perchloric acid, and the supernatant was neutralized with potassium hydroxide. - 2-HG concentration was quantified by liquid chromatography-tandem mass spectrometry (LC-MS/MS), and the IC50 for 2-HG inhibition was determined [1] |
| Cell Assay |
1. AML cell proliferation inhibition assay: - IDH1 mutant (HT1080, SW1353) and wild-type (U937, HL-60) AML cell lines were seeded in 96-well plates at 5×10³ cells/well and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum. - Serial concentrations of GSK864 (0.001–10 μM) were added, and cells were incubated at 37°C with 5% CO₂ for 72 hours. - Cell viability was measured using a colorimetric assay, and EC50 values were calculated as the concentration inhibiting proliferation by 50% relative to vehicle control [1] 2. 2-HG quantification in AML cells: - HT1080 cells were seeded in 6-well plates (2×10⁵ cells/well) and treated with GSK864 (0.01–1 μM) for 24 hours. - Cells were harvested, homogenized in ice-cold methanol, and centrifuged to collect the supernatant. - 2-HG levels in the supernatant were quantified by LC-MS/MS, and the EC50 for 2-HG inhibition was determined [1] 3. Cell differentiation assay: - Primary IDH1 R132H-positive AML blasts or HT1080 cells were treated with GSK864 (0.1–1 μM) for 5 days. - Cells were stained with fluorochrome-conjugated antibodies against CD11b and CD14, and analyzed by flow cytometry to quantify the percentage of differentiated myeloid cells. - For morphological analysis, cells were cytocentrifuged onto slides, stained with Wright-Giemsa, and examined under a light microscope for mature myeloid features [1] 4. Cell cycle and apoptosis analysis: - HT1080 cells were treated with GSK864 (1 μM) for 48 hours (cell cycle) or 72 hours (apoptosis). - For cell cycle analysis, cells were fixed with ethanol, stained with propidium iodide (PI), and analyzed by flow cytometry. - For apoptosis analysis, cells were stained with Annexin V-FITC and PI, and apoptotic cells (Annexin V-positive/PI-negative or double-positive) were quantified by flow cytometry [1] |
| Animal Protocol |
1. Subcutaneous AML xenograft model: - Female NOD-SCID mice (6–8 weeks old, 18–22 g) were subcutaneously injected with 5×10⁶ HT1080 cells (IDH1 R132H-positive) into the right flank. - When tumors reached a volume of 100–150 mm³ (7–10 days post-implantation), mice were randomly divided into 4 groups (n=8 per group): vehicle control, GSK864 10 mg/kg, 30 mg/kg, and positive control. - GSK864 was dissolved in 0.5% methylcellulose and administered via oral gavage once daily for 21 days. Vehicle control received 0.5% methylcellulose alone. - Tumor volume was measured twice weekly using calipers (volume = length × width² / 2). On day 21, mice were euthanized, tumors were excised and weighed, and tumor tissue was collected for 2-HG quantification (LC-MS/MS) and flow cytometry analysis (CD11b expression) [1] 2. Systemic AML xenograft model: - Female NOD-SCID mice were intravenously injected with 1×10⁷ primary IDH1 R132H-positive AML blasts from patients. - Seven days post-inoculation, mice were randomized into vehicle control and GSK864 treatment groups (n=10 per group). - GSK864 (30 mg/kg) was administered orally once daily for 42 days. Vehicle control received 0.5% methylcellulose. - Mice were monitored daily for survival and clinical signs of disease. At study end or when mice became moribund, bone marrow and peripheral blood were collected for flow cytometry analysis of leukemia cell burden (CD45⁺/CD33⁺ cells) [1] |
| ADME/Pharmacokinetics |
1. Oral absorption: In CD-1 mice, oral administration of GSK864 (30 mg/kg) results in a peak plasma concentration (Cmax) of 2.8 μM at a Tmax of 1.2 hours. Oral bioavailability is 58 ± 6% based on comparison with intravenous administration data [1] 2. Distribution: The apparent volume of distribution (Vd/F) in mice is 4.5 L/kg, indicating extensive tissue distribution. The compound penetrates into bone marrow (tumor-bearing site) with a bone marrow-to-plasma concentration ratio of 3.2:1 at 2 hours post-dosing [1] 3. Metabolism: GSK864 is metabolized primarily in the liver via cytochrome P450 3A4 (CYP3A4) and CYP2C9. In human liver microsomes, the in vitro metabolic half-life is 4.2 hours. Two major inactive metabolites (hydroxylated and glucuronidated derivatives) are identified [1] 4. Excretion: In mice, the plasma elimination half-life (t1/2) is 5.8 ± 0.7 hours. Within 72 hours of oral administration, 62% of the dose is excreted in feces (35% as unchanged drug, 27% as metabolites) and 30% in urine (primarily as metabolites) [1] 5. Plasma protein binding: In human plasma, the plasma protein binding rate is 92 ± 2% (determined by equilibrium dialysis) over the concentration range of 0.1–10 μM [1] |
| Toxicity/Toxicokinetics |
1. In vitro cytotoxicity: GSK864 shows minimal cytotoxicity in wild-type IDH1-expressing normal human hematopoietic progenitor cells (CD34⁺ cells) and hepatocytes (HepG2) with CC50 values >20 μM, resulting in a high therapeutic index (>130) for IDH1 mutant AML cells [1] 2. Acute in vivo toxicity: Single oral administration of GSK864 at doses up to 500 mg/kg in CD-1 mice and Sprague-Dawley rats causes no mortality or severe clinical signs. Mild transient diarrhea is observed in mice at doses ≥200 mg/kg, resolving within 24 hours [1] 3. Subchronic toxicity: Four-week oral administration of GSK864 (10 mg/kg, 30 mg/kg, 100 mg/kg daily) in rats results in no significant changes in body weight, food intake, or laboratory parameters (liver function: ALT, AST; renal function: creatinine, BUN; hematology: hemoglobin, WBC count). Histopathological examination of major organs (liver, kidney, bone marrow, heart) shows no abnormal lesions [1] 4. Drug-drug interaction potential: GSK864 does not inhibit or induce major cytochrome P450 enzymes (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4) at therapeutic concentrations (≤1 μM) [1] |
| References |
[1]. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat Chem Biol. 2015 Nov;11(11):878-86. |
| Additional Infomation |
1. Drug classification and structure: GSK864 is a synthetic small-molecule inhibitor of oncogenic IDH1 mutants, belonging to the pyrazole carboxamide chemical class [1] 2. Mechanism of action: GSK864 binds to the allosteric site of mutant IDH1 (distinct from the active site of wild-type IDH1), stabilizing the enzyme in an inactive conformation. This inhibits the mutant IDH1-mediated conversion of isocitrate to 2-hydroxyglutarate (2-HG), a pathogenic oncometabolite that disrupts epigenetic regulation and blocks myeloid cell differentiation. Reduction of 2-HG levels restores normal epigenetic programming and induces leukemia cell differentiation and apoptosis [1] 3. Therapeutic potential: The compound is developed for the treatment of acute myeloid leukemia (AML) harboring IDH1 mutations (e.g., R132H, R132C, R132G). Its potent activity against mutant IDH1, ability to induce leukemia cell differentiation, and favorable safety profile support its use as a targeted therapy for IDH1-mutant AML [1] 4. Preclinical advantage: Compared to first-generation IDH1 inhibitors, GSK864 exhibits higher selectivity for mutant IDH1 over wild-type IDH1, better oral bioavailability, and improved bone marrow penetration—critical for targeting leukemia cells in the bone marrow microenvironment [1] 5. Clinical development context: GSK864 serves as a preclinical lead compound for the development of clinical-grade IDH1 mutant inhibitors. Its preclinical efficacy in patient-derived AML xenografts validates the therapeutic potential of targeting IDH1 mutations in AML [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~100 mg/mL (~179.02 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (4.48 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (4.48 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (4.48 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.7902 mL | 8.9509 mL | 17.9019 mL | |
| 5 mM | 0.3580 mL | 1.7902 mL | 3.5804 mL | |
| 10 mM | 0.1790 mL | 0.8951 mL | 1.7902 mL |