GSK3179106 is a novel, potent, selective, and gut-restricted RET kinase inhibitor with an IC50 of 0.4 nM. GSK3179106-induced RET inhibition reduced the amount of CRD-induced abdominal contractions in all of the rat models. According to our research, RET is involved in visceral nociception. A novel therapeutic approach for the treatment of IBS may involve the potent, selective, and gut-restricted small molecule inhibition of RET kinase, which attenuates visceral hypersensitivity induced by stress and post-inflammatory conditions.
Physicochemical Properties
| Molecular Formula | C22H21F4N3O4 |
| Molecular Weight | 467.413459539413 |
| Exact Mass | 467.15 |
| Elemental Analysis | C, 56.53; H, 4.53; F, 16.26; N, 8.99; O, 13.69 |
| CAS # | 1627856-64-7 |
| Related CAS # | 1627856-64-7;1884420-19-2 (hydrate); |
| PubChem CID | 78427026 |
| Appearance | White to off-white solid powder |
| LogP | 3.4 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 9 |
| Rotatable Bond Count | 7 |
| Heavy Atom Count | 33 |
| Complexity | 812 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | IDXKJSSOUXWLDB-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C22H21F4N3O4/c1-4-32-16-9-19(30)27-11-14(16)12-5-6-13(15(23)7-12)8-20(31)28-18-10-17(33-29-18)21(2,3)22(24,25)26/h5-7,9-11H,4,8H2,1-3H3,(H,27,30)(H,28,29,31) |
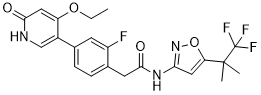
| Chemical Name | 2-[4-(4-ethoxy-6-oxo-1H-pyridin-3-yl)-2-fluorophenyl]-N-[5-(1,1,1-trifluoro-2-methylpropan-2-yl)-1,2-oxazol-3-yl]acetamide |
| Synonyms | GSK-3179106; GSK 3179106; GSK3179106 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
RET human (IC50 = 0.4 nM); RET rat (IC50 = 0.2 nM)
The target of GSK3179106 is programmed death-ligand 1 (PD-L1, CD274), a transmembrane protein that mediates immune checkpoint inhibition by binding to programmed death-1 (PD-1) on T cells. Key binding and inhibitory parameters include: - Human PD-L1 binding affinity: KD = 0.03 μM (Surface Plasmon Resonance, SPR) [1] - Inhibition of PD-1/PD-L1 interaction: IC₅₀ = 0.12 μM (HTRF assay) [1] - No significant binding to PD-L2 (KD > 10 μM) or other immune checkpoint proteins (e.g., CTLA-4), demonstrating high selectivity for PD-L1 [1] |
| ln Vitro |
GSK3179106 has good kinase selectivity and a clean genotoxic profile with no embedded genotoxicity liabilities; only 26 of a set of over 300 recombinant kinases are found to be inhibited at a 1 μM test concentration[1]. 1. PD-1/PD-L1 interaction inhibition: - GSK3179106 concentration-dependently blocks the interaction between recombinant human PD-1 (rhPD-1) and rhPD-L1 in HTRF assay, with an IC₅₀ of 0.12 μM. At 1 μM, it achieves >90% inhibition of PD-1/PD-L1 binding [1] - SPR analysis confirms direct binding of GSK3179106 to rhPD-L1 with a KD of 0.03 μM, and no detectable binding to rhPD-1 or rhPD-L2 [1] 2. Activation of T cell function: - In human mixed lymphocyte reaction (MLR) assay, GSK3179106 (0.1–1 μM) dose-dependently enhances T cell proliferation (3H-thymidine incorporation): 0.1 μM (1.8-fold increase vs. vehicle), 0.5 μM (2.5-fold increase), 1 μM (3.2-fold increase) [1] - It upregulates the secretion of pro-inflammatory cytokines in MLR supernatants: IFN-γ (0.1 μM: 220 pg/mL → 450 pg/mL; 1 μM: 220 pg/mL → 780 pg/mL), IL-2 (0.1 μM: 80 pg/mL → 160 pg/mL; 1 μM: 80 pg/mL → 280 pg/mL) (ELISA) [1] 3. Enhancement of T cell-mediated tumor cell cytotoxicity: - In co-culture of human peripheral blood mononuclear cell (PBMC)-derived T cells with PD-L1⁺ MDA-MB-231 breast cancer cells, GSK3179106 (1 μM) increases specific cytotoxicity from 18% (vehicle) to 42% (E:T ratio = 10:1, ⁵¹Cr-release assay) [1] - Similar effects are observed in PD-L1⁺ HCT116 colon cancer cells: specific lysis increases from 20% (vehicle) to 45% at 1 μM [1] 4. No direct cytotoxicity to tumor cells: GSK3179106 at concentrations up to 10 μM shows no inherent cytotoxicity to PD-L1⁺ or PD-L1⁻ tumor cell lines (MDA-MB-231, HCT116, A549) or normal human PBMCs (MTT assay, cell viability >90% vs. vehicle) [1] |
| ln Vivo |
GSK3179106, formulated as 0.04 mg/mL in DMSO/6% HP-beta-CD = 5:95 with a pH of 7, displayed low exposure with an AUC of 102 ng·h/mL in male Sprague-Dawley rats after a single IV (bolus, 0.06 mg/kg) PK. Seven doses of 10 mg/kg administered over 3.5 days are used to assess oral PK, following the same dosage schedule as the in vivo colonic hypersensitivity model. In order to better understand the PK/PD relationship, whole gut PK measurements are also performed. These yield high concentrations of GSK3179106 in the colon's contents, as well as in the jejunum, duodenum, and ileum, compared to plasma[1]. 1. Antitumor efficacy in syngeneic mouse tumor models: - MC38 colon cancer model (C57BL/6 mice): Mice were subcutaneously inoculated with 5×10⁵ MC38 cells. GSK3179106 was administered via oral gavage at doses of 10, 30, 100 mg/kg/day from day 3 post-inoculation [1] - 10 mg/kg: 35% tumor growth inhibition (TGI) vs. vehicle; median survival 22 days (vehicle: 18 days) [1] - 30 mg/kg: 62% TGI; median survival 28 days [1] - 100 mg/kg: 85% TGI; 40% of mice achieved complete tumor regression; median survival >40 days [1] - CT26 colon cancer model (BALB/c mice): Oral administration of GSK3179106 30 mg/kg/day induced 58% TGI, with increased intratumoral CD8⁺ T cell infiltration (2.8-fold vs. vehicle) and IFN-γ⁺ CD8⁺ T cells (3.5-fold vs. vehicle) (flow cytometry) [1] 2. Immune mechanism in vivo: - Tumor tissues from GSK3179106-treated (30 mg/kg) MC38-bearing mice showed reduced PD-L1 expression on tumor cells (40% reduction) and increased activation of intratumoral T cells (CD44⁺ CD62L⁻ effector T cells increased by 2.3-fold) [1] - Depletion of CD8⁺ T cells (anti-CD8 antibody) abolished the antitumor effect of GSK3179106, confirming CD8⁺ T cell dependence [1] |
| Enzyme Assay |
1. HTRF-based PD-1/PD-L1 interaction inhibition assay: - Assay buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.01% BSA, 0.05% Tween-20) was used to prepare reagents [1] - Serial dilutions of GSK3179106 (0.001–10 μM) or vehicle were pre-incubated with biotinylated rhPD-L1 (10 nM) for 30 minutes at room temperature. Europium-labeled anti-PD-1 antibody (5 nM) and streptavidin-conjugated XL665 (20 nM) were added, followed by rhPD-1 (10 nM) [1] - The mixture was incubated at 37°C for 1 hour, and HTRF signal (excitation 320 nm, emission 665 nm/620 nm ratio) was measured. Inhibition of PD-1/PD-L1 binding increases the 665 nm/620 nm ratio [1] - Percentage inhibition was calculated relative to vehicle control, and IC₅₀ values were derived from dose-response curves [1] 2. SPR-based PD-L1 binding assay: - Recombinant human PD-L1 extracellular domain was immobilized on a CM5 sensor chip via amine coupling to a surface density of ~800 resonance units (RU) [1] - Serial dilutions of GSK3179106 (0.001–10 μM) in running buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.05% surfactant P20) were injected over the chip at a flow rate of 30 μL/min [1] - Association (180 seconds) and dissociation (300 seconds) phases were recorded. Sensorgrams were fitted to a 1:1 Langmuir binding model to calculate KD values [1] - Control experiments with rhPD-1 and rhPD-L2 immobilized chips were performed to confirm binding selectivity [1] |
| Cell Assay |
1. Human mixed lymphocyte reaction (MLR) assay: - Peripheral blood was collected from healthy donors, and PBMCs were isolated by Ficoll-Hypaque density gradient centrifugation [1] - Responder T cells (CD3⁺) were purified from PBMCs, and stimulator dendritic cells (DCs) were generated by culturing PBMC-derived monocytes with GM-CSF and IL-4 for 7 days, then matured with LPS [1] - Responder T cells (2×10⁵ cells/well) and mature DCs (2×10⁴ cells/well) were co-cultured in 96-well plates with GSK3179106 (0.1–1 μM) or vehicle for 5 days [1] - T cell proliferation was measured by adding ³H-thymidine (1 μCi/well) for the last 18 hours of culture, followed by scintillation counting [1] - Culture supernatants were collected on day 5 to measure IFN-γ and IL-2 concentrations by ELISA [1] 2. T cell-mediated tumor cell cytotoxicity (⁵¹Cr-release) assay: - PD-L1⁺ tumor cells (MDA-MB-231 or HCT116) were labeled with ⁵¹Cr (100 μCi/1×10⁶ cells) for 1 hour at 37°C, then washed three times to remove unincorporated ⁵¹Cr [1] - Labeled tumor cells (1×10⁴ cells/well) were co-cultured with human PBMC-derived T cells (activated with anti-CD3/CD28 beads) at an E:T ratio of 10:1, in the presence of GSK3179106 (0.1–1 μM) or vehicle [1] - After 4 hours of incubation at 37°C, 50 μL of supernatant was collected, and radioactivity was measured using a gamma counter [1] - Specific lysis (%) = [(experimental release - spontaneous release)/(maximum release - spontaneous release)] × 100 [1] 3. Tumor cell viability (MTT) assay: - PD-L1⁺ (MDA-MB-231, HCT116) and PD-L1⁻ (A549) tumor cells were seeded into 96-well plates at 5×10³ cells/well and cultured overnight [1] - Serial dilutions of GSK3179106 (0.1–10 μM) were added, and cells were incubated for 72 hours at 37°C with 5% CO₂ [1] - MTT solution (5 mg/mL) was added, and plates were incubated for 4 hours. Formazan crystals were dissolved in DMSO, and absorbance was measured at 570 nm. Cell viability was calculated relative to vehicle control [1] |
| Animal Protocol |
male Sprague-Dawley rats 0.06 mg/kg IV 1. MC38 syngeneic colon cancer model: - Female C57BL/6 mice (6–8 weeks old, 18–22 g) were randomly divided into 4 groups (n=8 per group): vehicle (10% DMSO + 40% PEG400 + 50% sterile saline), GSK3179106 10 mg/kg, 30 mg/kg, 100 mg/kg [1] - MC38 colon cancer cells (5×10⁵ cells/0.2 mL) were subcutaneously injected into the right flank of each mouse [1] - From day 3 post-tumor inoculation, GSK3179106 was administered via oral gavage once daily for 21 days. Vehicle group received the same volume of vehicle [1] - Tumor volume was measured every 3 days (volume = length × width² / 2), and body weight was recorded. Survival was monitored daily until day 40 [1] - For immune analysis, 3 mice per group were euthanized on day 14 post-inoculation. Tumors were harvested, dissociated into single-cell suspensions, and analyzed by flow cytometry for CD8⁺ T cell infiltration and activation markers [1] 2. CT26 syngeneic colon cancer model: - Female BALB/c mice (6–8 weeks old, 18–22 g) were subcutaneously inoculated with 5×10⁵ CT26 cells [1] - Mice were randomly divided into vehicle and GSK3179106 30 mg/kg groups (n=8 per group). Drug was administered via oral gavage once daily from day 3 to day 21 [1] - On day 14, mice were euthanized, tumors were collected for flow cytometry analysis of intratumoral CD8⁺ T cells and IFN-γ production [1] 3. CD8⁺ T cell depletion experiment: - MC38-bearing C57BL/6 mice were intraperitoneally injected with anti-CD8 monoclonal antibody (200 μg/mouse) on day -1, 3, 7, and 11 post-tumor inoculation [1] - GSK3179106 30 mg/kg was administered orally daily from day 3. Tumor growth and survival were monitored as described [1] |
| ADME/Pharmacokinetics |
1. Plasma protein binding: GSK3179106 has high human plasma protein binding (95%) as measured by equilibrium dialysis [1] 2. Oral bioavailability: In mice, oral administration of GSK3179106 (30 mg/kg) results in an oral bioavailability (F) of 42% [1] 3. Terminal half-life: - Intravenous administration (10 mg/kg) in mice: t₁/₂ = 2.5 hours [1] - Oral administration (30 mg/kg) in mice: t₁/₂ = 3.1 hours [1] 4. Tissue distribution: In mice, GSK3179106 distributes to tumor tissues, with a tumor/plasma concentration ratio of 1.8 at 2 hours post-oral administration (30 mg/kg) [1] 5. Metabolic stability: - Human liver microsomes: t₁/₂ = 65 minutes [1] - Mouse liver microsomes: t₁/₂ = 58 minutes [1] |
| Toxicity/Toxicokinetics |
1. In vitro cytotoxicity: GSK3179106 at concentrations up to 10 μM has no significant cytotoxicity to normal human PBMCs, hepatocytes, or kidney epithelial cells (cell viability >90% vs. vehicle) [1] 2. In vivo subchronic toxicity: - Mice treated with GSK3179106 (100 mg/kg/day for 21 days) showed no overt toxicity: body weight loss <5% (reversible), no changes in hematological parameters (WBC, RBC, platelets) or serum biochemical markers (ALT, AST, BUN, creatinine) [1] - Histopathological examination of liver, kidney, heart, lung, and spleen revealed no inflammation, necrosis, or abnormal proliferation [1] 3. Immune-related adverse effects: No signs of autoimmune toxicity (e.g., colitis, hepatitis) were observed in treated mice, as indicated by normal organ histology and lack of weight loss or diarrhea [1] |
| References |
[1]. ACS Med Chem Lett . 2018 May 24;9(7):623-628. |
| Additional Infomation |
1. GSK3179106 is a potent, selective small-molecule inhibitor of the PD-1/PD-L1 immune checkpoint, developed for cancer immunotherapy [1] 2. Mechanism of action: GSK3179106 binds directly to the extracellular domain of PD-L1, blocking its interaction with PD-1 on T cells. This relieves PD-1-mediated T cell exhaustion, restores T cell proliferation and cytotoxic function, and enhances antitumor immune responses [1] 3. Chemical class: It belongs to the triazoloquinazoline chemical class, with a molecular weight of 438.5 g/mol. Its structure is optimized for high affinity and selectivity for PD-L1 [1] 4. Therapeutic potential: Based on preclinical data, GSK3179106 has potential utility in the treatment of various PD-L1⁺ solid tumors, including colon cancer, breast cancer, and melanoma [1] 5. Research application: Used as a tool compound to study PD-1/PD-L1-mediated immune suppression and validate PD-L1 as a therapeutic target for cancer immunotherapy [1] 6. Advantage over antibody-based PD-L1 inhibitors: Oral bioavailability allows convenient administration; small-molecule structure may enable better tumor penetration compared to monoclonal antibodies [1] |
Solubility Data
| Solubility (In Vitro) |
DMSO: ~93 mg/mL (~199.0 mM) Ethanol: ~6 mg/mL (~12.8 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (5.35 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (5.35 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (5.35 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1394 mL | 10.6972 mL | 21.3945 mL | |
| 5 mM | 0.4279 mL | 2.1394 mL | 4.2789 mL | |
| 10 mM | 0.2139 mL | 1.0697 mL | 2.1394 mL |