GSK2256098 is a small molecule developed by GlaxoSmithKline) as a selective FAK (Focal Adhesion Kinase) kinase inhibitor to inhibit FAK activity through targeting the phosphorylation site of FAK, tyrosine (Y) 397. The growth and survival of pancreatic ductal adenocarcinoma cells are inhibited by GSK2256098. When compared to Pyk2, the closest member of the FAK family, it is a thousand times more selective for FAK. By specifically targeting the phosphorylation site of FAK, tyrosine (Y) 397, GSK2256098 inhibits FAK activity. PTEN-mutant (Ishikawa) cells treated with GSK2256098 showed a higher degree of pFAK(Y397) inhibition than PTEN-wild-type (Hec1A) cells. Hec1A cells that had not been treated were less sensitive to GSK2256098 than Ishikawa cells. Ishikawa cells were transfected with a wild-type PTEN construct, and following GSK2256098 treatment, the expression of pFAK(Y397) remained unaltered. When paclitaxel and topotecan were combined with GSK2256098, Ishikawa cells showed decreased cell viability and increased sensitivity to the treatment, in contrast to Hec1a cells.
Physicochemical Properties
| Molecular Formula | C20H23CLN6O2 | |
| Molecular Weight | 414.89 | |
| Exact Mass | 414.157 | |
| Elemental Analysis | C, 57.90; H, 5.59; Cl, 8.54; N, 20.26; O, 7.71 | |
| CAS # | 1224887-10-8 | |
| Related CAS # | 1416771-10-2 (HCl);1224887-10-8; | |
| PubChem CID | 46214930 | |
| Appearance | White to off-white solid powder | |
| Density | 1.3±0.1 g/cm3 | |
| Boiling Point | 545.7±60.0 °C at 760 mmHg | |
| Flash Point | 283.8±32.9 °C | |
| Vapour Pressure | 0.0±1.5 mmHg at 25°C | |
| Index of Refraction | 1.638 | |
| LogP | 7.34 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 6 | |
| Rotatable Bond Count | 7 | |
| Heavy Atom Count | 29 | |
| Complexity | 539 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | ClC1=C([H])N=C(C([H])=C1N([H])C1=C([H])C([H])=C([H])C([H])=C1C(N([H])OC([H])([H])[H])=O)N([H])C1=C([H])C(C([H])([H])[H])=NN1C([H])(C([H])([H])[H])C([H])([H])[H] |
|
| InChi Key | BVAHPPKGOOJSPU-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C20H23ClN6O2/c1-12(2)27-19(9-13(3)25-27)24-18-10-17(15(21)11-22-18)23-16-8-6-5-7-14(16)20(28)26-29-4/h5-12H,1-4H3,(H,26,28)(H2,22,23,24) | |
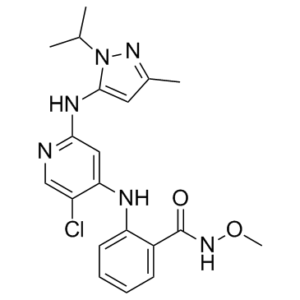
| Chemical Name | 2-[[5-chloro-2-[(5-methyl-2-propan-2-ylpyrazol-3-yl)amino]pyridin-4-yl]amino]-N-methoxybenzamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
FAK (Ki = 0.4 nM)
Focal Adhesion Kinase (FAK) (IC50 = 0.5 nM, recombinant FAK kinase assay) [1] Focal Adhesion Kinase (FAK) (IC50 = 0.7 nM, FAK autophosphorylation inhibition assay in Ishikawa cells; IC50 values for PTEN-deficient uterine cancer cells: 0.3-0.6 μM; PTEN-proficient cells: 1.2-1.8 μM) [2] |
| ln Vitro |
GSK2256098 is a thousand times more selective for FAK than Pyk2, the closest member of the FAK family. By specifically targeting the phosphorylation site of FAK, tyrosine (Y) 397, GSK2256098 inhibits FAK activity. At IC50s of 15, 8.5, and 12 nM, respectively, GSK2256098 inhibits FAK activity or Y397 phosphorylation in cancer cell lines OVCAR8 (ovary), U87MG (brain), and A549 (lung). Regarding FAK Y397 phosphorylation or activity, the responses of six PDAC cell lines to GSK2256098 treatments (0.1–10 μM) varied from low (less than 20% inhibition) to high (more than 90% inhibition). For additional examination, PANC-1 and L3.6P1, the least and most sensitive cell lines, are chosen. Reduced levels of phosphorylated Akt and ERK in L3.6P1 cells were correlated with GSK2256098 inhibition of FAK Y397 phosphorylation. Cell viability, anchorage-independent growth, and motility are all reduced by GSK2256098 in a dose-dependent way[1]. 1. Antiproliferative activity in pancreatic ductal adenocarcinoma (PDAC) cells: GSK2256098 dose-dependently inhibited the proliferation of PDAC cell lines (MIA PaCa-2, PANC-1, AsPC-1) with IC50 values of 0.4 μM, 0.7 μM, and 0.9 μM respectively after 72 hours of treatment (MTT assay). Normal pancreatic ductal epithelial cells (HPDE) showed lower sensitivity (IC50 > 5 μM) [1] 2. Antiproliferative activity in uterine cancer cells: GSK2256098 exhibited higher sensitivity in PTEN-deficient uterine cancer cells (Ishikawa, HEC-1A) with IC50 values of 0.3 μM and 0.6 μM (72 hours, CCK-8 assay), compared to PTEN-proficient cells (KLE, RL95-2) with IC50 values of 1.2 μM and 1.8 μM. PTEN knockdown in KLE cells enhanced sensitivity to GSK2256098 (IC50 reduced to 0.5 μM) [2] 3. FAK signaling pathway inhibition: GSK2256098 (0.1-1 μM) dose-dependently inhibited FAK autophosphorylation (Tyr397) in MIA PaCa-2 and Ishikawa cells, with 1 μM reducing p-FAK (Tyr397) levels by 80% and 75% respectively (Western blot). It also inhibited downstream signaling molecules, including p-Akt (Ser473) and p-ERK1/2, without affecting total FAK, Akt, or ERK1/2 expression [1][2] 4. Apoptosis induction: GSK2256098 (0.5-2 μM) induced apoptosis in MIA PaCa-2 and HEC-1A cells. Annexin V-FITC/PI staining showed apoptotic rates increased from 3-5% to 30-42% after 48 hours. Western blot revealed increased cleavage of PARP and caspase-3, and upregulation of Bax/Bcl-2 ratio [1][2] 5. Clonogenic inhibition: GSK2256098 (0.1-0.5 μM) dose-dependently inhibited colony formation of PANC-1 and Ishikawa cells. At 0.5 μM, colony formation rates were reduced by 75% (PANC-1) and 80% (Ishikawa) compared to the control group [1][2] 6. Inhibition of migration and invasion: GSK2256098 (0.3-1 μM) reduced the migration and invasion of MIA PaCa-2 cells (Transwell assay), with migration rate reduced by 55% (0.5 μM) and invasion rate reduced by 60% (0.5 μM). Similar effects were observed in HEC-1A cells, with migration and invasion rates reduced by 50% and 58% respectively at 0.5 μM [1][2] |
| ln Vivo |
The tumor samples taken from the therapy trials are analyzed because FAK is well-known to play a significant role in angiogenesis, proliferation, and apoptosis. By measuring CD31, tumors treated with GSK2256098 and Paclitaxel showed significantly lower microvessel densities than tumors from the vehicle control group (P<0.05). Ishikawa tumors exhibited the lowest microvessel density, although this was true for both models. In mice administered GSK2256098, all tumor models show reduced proliferation as measured by Ki67 compared to the control. The least responsive to therapy are Ishikawa tumors in terms of Ki67 expression. After receiving GSK2256098 treatment, Ishikawa tumors exhibit higher apoptotic indices than Hec1A tumors. All of the models that received treatment with the combination of GSK2256098 and Paclitaxel exhibit notable rates of apoptosis [2]. 1. PDAC xenograft model: Nude mice (BALB/c nu/nu) were subcutaneously implanted with MIA PaCa-2 cells. Oral administration of GSK2256098 (25, 50 mg/kg/day) for 28 days dose-dependently inhibited tumor growth, with tumor growth inhibition (TGI) rates of 45% (25 mg/kg) and 70% (50 mg/kg) compared to the vehicle group. The 50 mg/kg group significantly prolonged median from 42 days (vehicle) to 65 days. Tumor tissues showed reduced p-FAK (Tyr397) expression by 68% and decreased Ki-67 proliferation index (from 70% to 25%) [1] |
| Enzyme Assay |
The closest member of the FAK family, Pyk2, is a thousand times less selective for FAK than GSK2256098. By focusing on FAK's phosphorylation site, tyrosine (Y) 397, GSK2256098 suppresses FAK activity. For OVCAR8 (ovary), U87MG (brain), and A549 (lung) cancer cell lines, GSK2256098 inhibits FAK activity or Y397 phosphorylation at IC50s of 15, 8.5, and 12 nM, respectively. 1. Recombinant FAK kinase assay: Recombinant human FAK protein was incubated with different concentrations of GSK2256098 (0.01-10 nM) and a synthetic peptide substrate containing the FAK phosphorylation site in kinase buffer. The reaction was initiated by adding ATP (5 μM) and incubated at 30℃ for 60 minutes. The phosphorylated substrate was detected using a homogeneous time-resolved fluorescence (HTRF) assay, and the inhibition rate was calculated to determine the IC50 value [1] 2. FAK autophosphorylation inhibition assay: Ishikawa cells were seeded in 24-well plates and serum-starved for 24 hours. GSK2256098 (0.01-10 nM) was added, and cells were incubated for 1 hour, then stimulated with fibronectin (10 μg/mL) for 30 minutes to induce FAK autophosphorylation. Cells were lysed, and p-FAK (Tyr397) levels were detected by Western blot. The IC50 for inhibiting FAK autophosphorylation was calculated from dose-response curves [2] |
| Cell Assay |
On the wells of a 96-well plate, PDAC cells are grown. The wells are filled with 10 microliters of MTS (100 μL total). The absorbance at 450 nm wave length of reacted MTS is measured on a microplate reader after the plate is incubated for 10 to 30 minutes at 37°C. GSK2256098's IC50 on cell viability is determined using the Sigma plot program. Using a 6-well plate, PDAC cells are grown. The cells are incubated in the medium containing 0.1–10 μM GSK2256098 for 48 or 72 hours after cell confluence in regular medium reaches roughly 70%. Cells are reseeded at the conclusion of treatments and stored for nine days. The blue colonies are then counted after the cells are stained with clonogenic reagent[1]. Cell viability assay [1] PDAC cells were cultured on the wells of a 96-well plate. Ten microliters of MTS was added to the wells (total value: 100 μl). After the plate was kept in a 37°C incubator for 10–30 min, the absorbance at 450 nm wave length of reacted MTS was determined on a microplate reader. The Sigma plot program was used to calculate IC50 of GSK2256098 on cell viability. Cell clonogenic survival assay [1] PDAC cells were cultured on a 6-well plate. When cell confluence reached about 70% in regular medium, the cells were incubated in the medium containing 0.1–10 μM GSK2256098 for 48 or 72 hr. At the end of treatments, cells were re-seeded and kept for 9 d Then, the cells were stained using Clonogenic Reagent, and the blue colonies were counted. Soft agar assay [1] Soft agar assay was performed to assess anchorage-independent growth of the pancreatic cancer cells. Briefly, 1.5 ml of media was combined with 1.5 ml of 1% bact-agar solution in dishes and allowed to solidify, creating the bottom layer of agar-growth media. The cells in growth media containing agarose solution and 0–25 μM GSK2256098 were plated on top of the medium-agar layer. The dishes containing agarose-suspended cells were kept in a 37°C CO2 incubator for 2–4 weeks with regular cell feeding (twice per week). The cells were stained using Crystal Violet. Plates were divided into quadrants. Colonies were counted in the whole plate and normalized to control cells. Cell motility analysis [1] We used a wound healing assay to assess the effects of GSK2256098 on cell motility. PDAC cells were cultured on the wells of a 6-well plate. When cells reached confluence, a straight scratch was made using a yellow sterile tip. Cells were cultured in regular medium containing 0–10 μM GSK2256098. Micro-images of the scratches were taken under a microscope and used as control (0 hr). After the plate was kept for 48 hr, the micro-images of the gaps were taken. The distance of the gaps were measured, and percentages of GSK2256098-inhibited wound healing were calculated. Cell viability assay [2] To test the sensitivity of Ishikawa and Hec1A cells to treatment, 2,000 cells per well were plated into a 96-well plate and allowed to adhere overnight. After 12 hours of serum deprivation, they were treated in triplicate with GSK2256098 at increasing concentrations (0.01-10 μmol/L) in medium without serum. After 24 hours of treatment, cell viability was assessed by adding 50 μL of 0.15% 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide to each well. After two hours of incubation at 37°C, medium/3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was removed, 200 μL dimethyl sulfoxide was added to each well, and the absorbance at 570 nm was recorded using a Falcon plate reader. The cell viability was determined by calculating the mean absorbance at 570 nm into a percentage from 100% of the untreated cells’ mean absorbance, as previously described. For combined GSK2256098-based treatment and chemotherapy, 1 μM GSK2256098 and a range of paclitaxel, cisplatin, or topotecan doses were tested. 1. Cell proliferation assay: PDAC cells (MIA PaCa-2, PANC-1) and uterine cancer cells (Ishikawa, HEC-1A, KLE) were seeded in 96-well plates at a density of 2×10^3 cells/well. After 24 hours of adherence, cells were treated with GSK2256098 (0.01-10 μM) for 72 hours. MTT (for PDAC cells) or CCK-8 (for uterine cancer cells) reagent was added, and after 4 hours of incubation, absorbance at 570 nm (MTT) or 450 nm (CCK-8) was measured to calculate cell viability and IC50 values [1][2] 2. Western blot assay: Cells or tumor tissues were lysed in RIPA buffer containing protease and phosphatase inhibitors. Total protein was separated by SDS-PAGE, transferred to PVDF membranes, and probed with antibodies against p-FAK (Tyr397), FAK, p-Akt (Ser473), Akt, p-ERK1/2, ERK1/2, cleaved PARP, cleaved caspase-3, Bax, Bcl-2, Ki-67, and GAPDH. Chemiluminescent signals were detected and quantified [1][2] 3. Apoptosis assay: MIA PaCa-2 and HEC-1A cells were seeded in 6-well plates (5×10^5 cells/well) and treated with GSK2256098 (0.5, 1, 2 μM) for 48 hours. Cells were harvested, stained with Annexin V-FITC and PI, and analyzed by flow cytometry to quantify apoptotic cells [1][2] 4. Clonogenic assay: PANC-1 and Ishikawa cells were seeded in 6-well plates (200 cells/well) and allowed to adhere for 24 hours. GSK2256098 (0.1, 0.3, 0.5 μM) was added, and cells were cultured for 14 days. Colonies were fixed with methanol, stained with crystal violet, and counted. The colony formation rate was calculated as (number of colonies in treatment group / number in control group) × 100% [1][2] 5. Transwell migration and invasion assay: MIA PaCa-2 and HEC-1A cells were resuspended in serum-free medium and seeded in the upper chamber of transwell inserts (8 μm pore size) for migration assay, or Matrigel-coated inserts for invasion assay (5×10^4 cells/well). GSK2256098 (0.3, 0.5, 1 μM) was added to both chambers, and the lower chamber contained medium with 10% fetal bovine serum. After 24 hours (migration) or 48 hours (invasion), cells were fixed, stained, and counted under a microscope [1][2] |
| Animal Protocol |
Mice: The mice used are female athymic nude mice aged 8–12 weeks. In experimental therapy, 4x106 Ishikawa or Hec1A cells are injected into the uterus horn. The mice are randomized (n = 10 mice per group) based on the following groups after receiving tumor cell injection: 1) 100 microliters of a vehicle control (orally, daily); 2) 75 milligrams per kilogram of GSK2256098 in 100 microliters of vehicle (orally, daily); 3) 2.5 milligrams per kilogram of Paclitaxel in 200 microliters of PBS (intraperitoneally, weekly); and 4) GSK2256098 and Paclitaxel (doses and frequencies mentioned above). After the tumor injection, therapy is started 10–14 days later. Four to six weeks after treatment initiation, the mice are checked for side effects and cervically dislocated for death. Each mouse's weight, the total weight of the tumor, the location and quantity of tumor nodules are noted for each treatment group at the end of the experiment. In order to process tumor samples for additional analysis, they are either frozen in a medium with the ideal cutting temperature or paraffin-embedded in a section fixed in formalin. 1. PDAC xenograft model: Female BALB/c nu/nu mice (6-8 weeks old, 18-22 g) were subcutaneously injected with 5×10^6 MIA PaCa-2 cells mixed with Matrigel (1:1 ratio) into the right flank. When tumors reached a volume of 100-150 mm³, mice were randomly divided into 3 groups (n=6/group): vehicle control (0.5% methylcellulose), GSK2256098 25 mg/kg, and 50 mg/kg. The drug was suspended in 0.5% methylcellulose and administered orally by gavage once daily for 28 days. Tumor volume was measured every 3 days (calculated as length × width² / 2), and body weight was recorded daily. At the end of the experiment, mice were sacrificed, tumors were excised, weighed, and stored for Western blot and immunohistochemical analysis [1] |
| ADME/Pharmacokinetics |
1. Oral bioavailability: In rats, oral administration of GSK2256098 (10 mg/kg) resulted in an absolute bioavailability of 42% [1] 2. Plasma pharmacokinetics: After oral administration (10 mg/kg) in rats, the peak plasma concentration (Cmax) was 1.3 μM (achieved at 1.5 hours), area under the curve (AUC0-24h) was 9.8 μM·h, and elimination half-life (t1/2) was 6.5 hours [1] 3. Tissue distribution: In mice, 2 hours after oral administration of GSK2256098 (50 mg/kg), the highest drug concentrations were detected in the liver (5.2 μM) and tumor tissue (3.1 μM), followed by the kidney (2.8 μM) and lung (1.6 μM). Brain concentration was undetectable (<0.1 μM) [1] |
| Toxicity/Toxicokinetics |
1. Acute toxicity: In rats, single oral administration of GSK2256098 at doses up to 200 mg/kg did not cause significant mortality or obvious toxic symptoms (e.g., lethargy, diarrhea, weight loss) within 14 days of observation [1] 2. Chronic toxicity: Mice treated with GSK2256098 (50 mg/kg/day, oral) for 28 days showed no significant changes in liver function (ALT, AST) or kidney function (BUN, creatinine) compared to the vehicle group. Histopathological analysis of major organs (liver, kidney, heart, lung) revealed no abnormal lesions [1] |
| References |
[1]. A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell Cycle. 2014;13(19):3143-9. [2]. PTEN Expression as a Predictor of Response to Focal Adhesion Kinase Inhibition in Uterine Cancer. Mol Cancer Ther. 2015 Jun;14(6):1466-75. |
| Additional Infomation |
GSK2256098 is under investigation in clinical trial NCT02523014 (Vismodegib and FAK Inhibitor GSK2256098 in Treating Patients With Progressive Meningiomas). FAK Inhibitor GSK2256098 is a focal adhesion kinase-1 (FAK) inhibitor with potential antiangiogenic and antineoplastic activities. FAK inhibitor GSK2256098 inhibits FAK, which may prevent the integrin-mediated activation of several downstream signal transduction pathways, including ERK, JNK/MAPK and PI3K/Akt, thereby inhibiting tumor cell migration, proliferation and survival, and tumor angiogenesis. The tyrosine kinase FAK is normally activated by binding to integrins in the extracellular matrix (ECM) but may be upregulated and constitutively activated in various tumor cell types. Focal adhesion kinase (FAK) hyperactivation is common in pancreatic ductal adenocarcinoma (PDAC). A small molecule, GSK2256098 (GlaxoSmithKline), has been developed to inhibit FAK activity through targeting the phosphorylation site of FAK, tyrosine (Y) 397. We sought to determine whether GSK2256098 inhibition of FAK Y397 phosphorylation attenuates PDAC-associated cell proliferation, motility and survival. Cultured PDAC cells were used as cellular models of GSK2256098-impaired abnormal growth. Western blot analysis, cell viability analysis, clonogenic survival, soft-agar and wound healing assays were performed. The responses of 6 PDAC cell lines in regards to FAK Y397 phosphorylation or activity to GSK2256098 treatments (0.1-10 μM) ranged from low (less than 20% inhibition) to high (more than 90% inhibition). The least and most sensitive cell lines (PANC-1 and L3.6P1) were selected for further analysis. GSK2256098 inhibition of FAK Y397 phosphorylation correlated with decreased levels of phosphorylated Akt and ERK in L3.6P1 cells. GSK2256098 decreased cell viability, anchorage-independent growth, and motility in a dose dependent manner. Current studies demonstrate that small molecule kinase inhibitors targeting FAK Y397 phosphorylation can inhibit PDAC cell growth. Assessments of FAK Y397 phosphorylation in biopsies may be used as a biomarker to select the subgroup of responsive patients and/or monitor the effects of GSK2256098 on FAK-modulated tumor growth during treatment.[1] PTEN is known to be frequently mutated in uterine cancer and also dephosphorylates FAK. Here, we examined the impact of PTEN alterations on the response to treatment with a FAK inhibitor (GSK2256098). In vitro and in vivo therapeutic experiments were carried out using PTEN-mutated and PTEN-wild-type models of uterine cancer alone and in combination with chemotherapy. Treatment with GSK2256098 resulted in greater inhibition of pFAK(Y397) in PTEN-mutated (Ishikawa) than in PTEN-wild-type (Hec1A) cells. Ishikawa cells were more sensitive to GSK2256098 than the treated Hec1A cells. Ishikawa cells were transfected with a wild-type PTEN construct and pFAK(Y397) expression was unchanged after treatment with GSK2256098. Decreased cell viability and enhanced sensitivity to chemotherapy (paclitaxel and topotecan) in combination with GSK2256098 was observed in Ishikawa cells as compared with Hec1a cells. In the Ishikawa orthoptopic murine model, treatment with GSK2256098 resulted in lower tumor weights and fewer metastases than mice inoculated with Hec1A cells. Tumors treated with GSK2256098 had lower microvessel density (CD31), less cellular proliferation (Ki67), and higher apoptosis (TUNEL) rates in the Ishikawa model when compared with the Hec1a model. From a large cohort of evaluable patients, increased FAK and pFAK(Y397) expression levels were significantly related to poor overall survival. Moreover, PTEN levels were inversely related to pFAK(Y397) expression. These preclinical data demonstrate that PTEN-mutated uterine cancer responds better to FAK inhibition than does PTEN wild-type cancer. Therefore, PTEN could be a biomarker for predicting response to FAK-targeted therapy during clinical development.[2] 1. GSK2256098 is a potent and selective small-molecule inhibitor of FAK, a non-receptor tyrosine kinase that regulates cell proliferation, survival, migration, and invasion through downstream PI3K/Akt and MAPK/ERK signaling pathways. Its mechanism of action involves binding to the ATP-binding pocket of FAK, inhibiting its autophosphorylation (Tyr397) and subsequent activation of downstream signaling [1][2] 2. In uterine cancer cells, GSK2256098 exhibits higher efficacy in PTEN-deficient cells compared to PTEN-proficient cells, suggesting that PTEN expression status may serve as a predictive biomarker for response to FAK inhibition. PTEN deficiency leads to increased FAK activation, making cells more dependent on FAK signaling for survival [2] 3. GSK2256098 shows significant antitumor activity in preclinical models of PDAC, including inhibition of tumor growth and prolongation of survival, with favorable safety profiles. Its ability to inhibit cancer cell migration and invasion also suggests potential for preventing metastasis. The drug has moderate oral bioavailability and effective tumor tissue penetration, supporting its potential as a targeted therapy for FAK-dependent cancers [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (6.03 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (6.03 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4103 mL | 12.0514 mL | 24.1028 mL | |
| 5 mM | 0.4821 mL | 2.4103 mL | 4.8206 mL | |
| 10 mM | 0.2410 mL | 1.2051 mL | 2.4103 mL |