Physicochemical Properties
| Molecular Formula | C17H19N5O2S |
| Molecular Weight | 357.430061578751 |
| Exact Mass | 357.125 |
| CAS # | 1257705-09-1 |
| PubChem CID | 67479440 |
| Appearance | White to off-white solid powder |
| LogP | 1 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 3 |
| Heavy Atom Count | 25 |
| Complexity | 544 |
| Defined Atom Stereocenter Count | 0 |
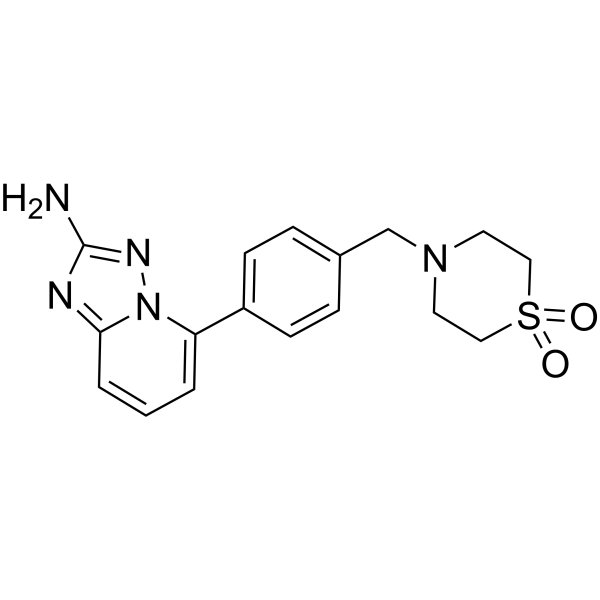
| SMILES | S1(CCN(CC2C=CC(C3=CC=CC4=NC(N)=NN34)=CC=2)CC1)(=O)=O |
| InChi Key | BYWJAVQGRJEEHH-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C17H19N5O2S/c18-17-19-16-3-1-2-15(22(16)20-17)14-6-4-13(5-7-14)12-21-8-10-25(23,24)11-9-21/h1-7H,8-12H2,(H2,18,20) |
| Chemical Name | 5-[4-[(1,1-dioxo-1,4-thiazinan-4-yl)methyl]phenyl]-[1,2,4]triazolo[1,5-a]pyridin-2-amine |
| Synonyms | 1257705-09-1; [1,2,4]Triazolo[1,5-a]pyridin-2-amine, 5-[4-[(1,1-dioxido-4-thiomorpholinyl)methyl]phenyl]-; GS-829845; 5-[4-[(1,1-dioxo-1,4-thiazinan-4-yl)methyl]phenyl]-[1,2,4]triazolo[1,5-a]pyridin-2-amine; 5-[4-(1,1-Dioxothiomorpholin-4-ylmethyl)-phenyl]-[1,2,4]triazolo[1,5-a]pyridin-2-ylamine; CHEMBL3360840; 4-(4-(2-Amino-[1,2,4]triazolo[1,5-a]pyridin-5-yl)benzyl)thiomorpholine 1,1-dioxide; 5-{4-[(1,1-dioxidothiomorpholin-4-yl)methyl]phenyl}[1,2,4]triazolo[1,5-a]pyridin-2-amine; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment (e.g. under nitrogen), avoid exposure to moisture and light. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Active/major metabolite of Filgotinib |
| ln Vitro | GS-829845 is a major and active metabolite of Filgotinib. As a JAK1 inhibitor, GS-829845 is about 10-fold less potent than the parent filgotinib but has a longer half-life than filgotinib. |
| ln Vivo | The nonlinear logistic regression showed increasing response with increasing exposure, with exposures at 200 mg dose primarily residing on the curve plateau. Also, AUCeff was significantly higher in the subjects who achieved responses compared to those who did not (10 900 vs 9900 h*ng/mL for ACR20, P value < .0001). For exposure-safety analyses, filgotinib and GS-829845 exposures were similar irrespective of the presence/absence of the evaluated safety endpoints, indicating no exposure-safety relationship for common treatment-emergent adverse events (TEAEs)/laboratory abnormalities and serious TEAEs/infections. Conclusions: ER analyses confirmed that filgotinib produced more robust therapeutic effects across the exposure range observed at 200 mg once daily compared to lower doses, and collectively with the lack of exposure-safety relationship, the 200 mg once daily dose was supported for commercialization.[1] |
| Enzyme Assay | Filgotinib, a preferential Janus Kinase-1 inhibitor, is approved in Europe and Japan for treatment of rheumatoid arthritis and is being developed for treatment of other chronic inflammatory diseases. Three drug-drug interactions studies were conducted in healthy subjects to evaluate the effect of P-glycoprotein (P-gp) modulation (study 1: P-gp inhibition by itraconazole and study 2: P-gp induction by rifampin) on filgotinib pharmacokinetics and the potential of filgotinib to impact exposure of metformin, an organic cation transporter (OCT) 2 and multidrug and toxin extrusion (MATE) 1/2K substrate (study 3). Co-administration of filgotinib with itraconazole increased filgotinib exposure (maximum concentration [Cmax ] by 64% and area under the curve to infinity [AUCinf ] by 45%) but had no effect on the exposure of GS-829845, filgotinib's primary metabolite. Rifampin moderately reduced exposures of filgotinib and GS-829845 (Cmax by 26% and AUCinf by 27% for filgotinib; Cmax by 19% and AUCinf by 38% for GS-829845). The data confirmed that filgotinib is a P-gp substrate. However, the magnitude of change in filgotinib/GS-829845 exposure by P-gp modulators is not deemed to be clinically relevant based on filgotinib exposure-response analyses in subjects with rheumatoid arthritis. Filgotinib did not alter metformin exposures, indicating that filgotinib and GS-829845 do not inhibit OCT2 and MATE1/2K at the clinical doses. Filgotinib was generally well-tolerated when administered alone or with the co-administered drugs in the studies. Results from these studies were the basis to enable the use of P-gp modulators and substrates of OCT2, MATE1, and MATE2K with filgotinib without the need for dose modifications in the current approved rheumatoid arthritis population.[2] |
| Animal Protocol | Filgotinib is a potent, oral, JAK1-preferential inhibitor for the treatment of rheumatoid arthritis (RA). This report describes exposure-response (ER) analyses of filgotinib for dose confirmation based on three phase 3 and two phase 2 studies in moderate to severe RA patients. Methods: The pharmacokinetic exposures used in ER analyses were derived from population pharmacokinetic analysis. The exposure-efficacy relationships were assessed for efficacy endpoints (ACR20/50/70 and DAS28) over effective area under curve (AUCeff ), the combined exposures of filgotinib and GS-829845 (major, active metabolite), with nonlinear logistic regression models developed. Also, a t-test was performed to compare the exposure between subjects who achieved response and those who did not. For the ER analyses of safety, exposures were examined between subjects who experienced and who did not experience the evaluated safety events, which was conducted separately for filgotinib and GS-829845.[1] |
| References |
[1]. Exposure-response relationships for the efficacy and safety of filgotinib and its metabolite GS-829845 in subjects with rheumatoid arthritis based on phase 2 and phase 3 studies. Br J Clin Pharmacol. 2022 Jul;88(7):3211-3221. [2]. Evaluation of the potential drug interactions mediated through P-gp, OCT2, and MATE1/2K with filgotinib in healthy subjects. Clin Transl Sci. 2022 Feb;15(2):361-370. |
Solubility Data
| Solubility (In Vitro) | DMSO : ~100 mg/mL (~279.78 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (6.99 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: 2.5 mg/mL (6.99 mM) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (6.99 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7978 mL | 13.9888 mL | 27.9775 mL | |
| 5 mM | 0.5596 mL | 2.7978 mL | 5.5955 mL | |
| 10 mM | 0.2798 mL | 1.3989 mL | 2.7978 mL |