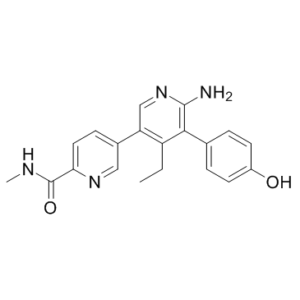
GNE-6776 (GNE6776) is a novel, highly potent, specific, non-covalent, and orally bioavailable USP7 (Ubiquitin specific proteases7) inhibitor with anticancer activity. It is more selective for USP7 (IC50 of ~1.34 uM) than USP47 and USP5 (IC50>200 uM). GNE-6776 can also promote endogenous MDM2 ubiquitination, stabilize p53 and upregulate p21, induce tumour cell death and enhance cytotoxicity with chemotherapeutic agents and targeted compounds such as PIM kinase inhibitors; GNE-6776 promotes on-target pathway modulation in human xenografts. Structural studies reveal that GNE-6776 non-covalently targets USP7 12 Å distant from the catalytic cysteine. GNE-6776 attenuates ubiquitin binding and thus inhibits USP7 deubiquitinase activity. It interacts with acidic residues that mediate hydrogen-bond interactions with the ubiquitin Lys48 side chain, suggesting that USP7 preferentially interacts with and cleaves ubiquitin moieties that have free Lys48 side chains.
Physicochemical Properties
| Molecular Formula | C20H20N4O2 | |
| Molecular Weight | 348.398404121399 | |
| Exact Mass | 348.15862 | |
| CAS # | 2009273-71-4 | |
| Related CAS # |
|
|
| PubChem CID | 122531750 | |
| Appearance | White to off-white solid powder | |
| LogP | 2.7 | |
| Hydrogen Bond Donor Count | 3 | |
| Hydrogen Bond Acceptor Count | 5 | |
| Rotatable Bond Count | 4 | |
| Heavy Atom Count | 26 | |
| Complexity | 467 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | OC1C=CC(=CC=1)C1C(N)=NC=C(C2C=NC(C(NC)=O)=CC=2)C=1CC |
|
| InChi Key | UCYSSYGGXOFJKK-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C20H20N4O2/c1-3-15-16(13-6-9-17(23-10-13)20(26)22-2)11-24-19(21)18(15)12-4-7-14(25)8-5-12/h4-11,25H,3H2,1-2H3,(H2,21,24)(H,22,26) | |
| Chemical Name | 5-[6-amino-4-ethyl-5-(4-hydroxyphenyl)pyridin-3-yl]-N-methylpyridine-2-carboxamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
USP7
The target of GNE-6776 is Ubiquitin-Specific Peptidase 7 (USP7), a deubiquitinase enzyme. It non-covalently binds to USP7 at a site 12 Å away from the catalytic cysteine, interfering with ubiquitin binding to USP7 and thereby inhibiting USP7's deubiquitinase activity. Specific IC50, Ki, or EC50 values for the inhibition of USP7 by GNE-6776 are not explicitly provided in the referenced literature [1] |
| ln Vitro |
At 15 μM, GNE-6776 significantly inhibits USP7. A highly selective USP7 inhibitor against endogenous cellular deubiquitinases as well as recombinant ones is GNE-6776[1]. GNE-6776 induced tumour cell death and enhance cytotoxicity with chemotherapeutic agents and targeted compounds, including PIM kinase inhibitors. Structural studies reveal that GNE-6640 and GNE-6776 non-covalently target USP7 12 Å distant from the catalytic cysteine. The compounds attenuate ubiquitin binding and thus inhibit USP7 deubiquitinase activity. GNE-6640 and GNE-6776 interact with acidic residues that mediate hydrogen-bond interactions with the ubiquitin Lys48 side chain, suggesting that USP7 preferentially interacts with and cleaves ubiquitin moieties that have free Lys48 side chains. 1. GNE-6776 induces death of tumor cells in vitro. Although specific tumor cell lines and quantitative data (such as cell viability rates at different concentrations) are not detailed, the compound shows cytotoxic effects on tumor cells, which is associated with its inhibition of USP7 activity [1] 2. GNE-6776 enhances the cytotoxicity of chemotherapeutic agents and targeted compounds in vitro, including PIM kinase inhibitors. When used in combination with these agents, the inhibitory effect on tumor cell survival is more significant compared to the use of GNE-6776 alone or the other agents alone, indicating a synergistic effect [1] 3. GNE-6776 attenuates ubiquitin binding to USP7, thereby inhibiting USP7's deubiquitinase activity. Structural studies confirm that GNE-6776 interacts with acidic residues in USP7 that mediate hydrogen-bond interactions with the ubiquitin Lys48 side chain. This interaction disrupts the binding between USP7 and ubiquitin, especially affecting USP7's preference for Lys48-linked ubiquitin chains [1] 4. GNE-6776 protracts the depolymerization kinetics of Lys48-linked ubiquitin chains by USP7 relative to Lys63-linked chains in vitro. This was demonstrated by engineering di-ubiquitin chains with differential proximal and distal isotopic labels and measuring USP7 binding via nuclear magnetic resonance (NMR). The results show that USP7's ability to depolymerize Lys48-linked chains is more significantly inhibited by GNE-6776 [1] |
| ln Vivo |
GNE-6776 inhibits the growth of EOL-1 xenografts in mice when given orally once or twice a day for ten days at a dose of 100 or 200 mg/kg. [1]. Given the favourable features of these inhibitors, we investigated their efficacy in animal models. Pharmacodynamic and pharmacokinetic studies indicated that GNE-6776 is orally bioavailable and promotes on-target pathway modulation in human xenografts (Extended Data Fig. 4e–i). Although efficacious exposure was only transiently achieved, GNE-6776 caused modest, although significant, EOL-1 xenograft growth delay (Extended Data Fig. 4j). Developing USP7 inhibitors that have improved drug-like properties will be necessary to comprehensively evaluate USP7 inhibition in vivo.[1] |
| Enzyme Assay |
USP7 enzymatic analysis[1] Michaelis–Menten kinetic measurements with full-length USP7 were performed using 1 nM USP7 with a series of ubiquitin–AMC substrate titrations. The initial rate of substrate hydrolysis was determined using the Magellan software on a Tecan Safire2 plate reader, and kinetic parameters modelled using nonlinear regression analysis with GraphPad Prism software. Standard error was calculated from three technical replicates. For studies using the USP7 D305/E308 mutant, samples were reacted in a buffer consisting of 50 mM HEPES (pH 7.5), 100 mM NaCl, 2.5 mM dithiothreitol, and 0.1% (w/v) bovine gamma globulin. The starting substrate concentration of ubiquitin-Rho110 used for the Michaelis–Menten analysis was 100 μM serial diluted to 781 nM. Reactions were performed for 1 h at room temperature with a final enzyme concentration of 100 nM (three independent experiments, see symbols in plots), in black 100-μl volume 96-well half area plates. The enzymatic activity was calculated by fitting the data using the initial velocity with the linear V0 values measured by analysing the fluorescence signal of cleaved Rho-110 using excitation at 485 nm and emission at 535 nm. Deubiquitinase selectivity analysis[1] Recombinant deubiquitinase di-ubiquitin mass spectrometry cleavage assay. The MALDI–TOF DUB assay was performed using the indicated concentrations of recombinant deubiquitinases, di-ubiquitin substrates, and USP7 inhibitor compounds as described previously. The inhibition efficiency for GNE-6640 and GNE-6776 against the UCHl family members was monitored on Ub-Ube2W (Ub-E2), an alternative substrate to di-ubiquitin. 1. NMR-based USP7-ubiquitin binding assay: To assess the effect of GNE-6776 on USP7-ubiquitin binding, nuclear magnetic resonance (NMR) spectroscopy was used. First, appropriate samples containing USP7 (or its relevant domains) and ubiquitin were prepared. Then, GNE-6776 at different concentrations was added to the samples. NMR spectra of the samples were collected and analyzed to monitor changes in the interaction between USP7 and ubiquitin. The results revealed that GNE-6776 interferes with the binding of USP7 to ubiquitin, as indicated by changes in relevant NMR signals [1] 2. USP7 deubiquitinase activity assay: To measure the inhibitory effect of GNE-6776 on USP7's deubiquitinase activity, a reaction system containing USP7 enzyme, ubiquitin-substrate (such as di-ubiquitin chains with specific linkages like Lys48-linked or Lys63-linked chains), and appropriate buffers was established. GNE-6776 at different concentrations was added to the reaction system, and the mixture was incubated under suitable conditions (e.g., specific temperature and time). After incubation, the reaction products were analyzed using appropriate methods (such as gel electrophoresis or mass spectrometry) to determine the extent of ubiquitin chain depolymerization. The assay showed that GNE-6776 inhibits USP7's deubiquitinase activity, particularly affecting the depolymerization of Lys48-linked ubiquitin chains [1] 3. Isotopically labeled di-ubiquitin chain binding assay: To investigate USP7's preference for different ubiquitin chain linkages and the effect of GNE-6776 on this preference, di-ubiquitin chains with differential proximal and distal isotopic labels (e.g., ¹³C/¹⁵N labeling) were engineered. These labeled chains (including Lys48-linked and Lys63-linked types) were incubated with USP7 in the presence or absence of GNE-6776. NMR spectroscopy was used to measure and compare USP7's binding to the different labeled chains. This assay confirmed that USP7 preferentially interacts with ubiquitin moieties having free Lys48 side chains, and GNE-6776 attenuates this preferential binding, leading to prolonged depolymerization kinetics of Lys48-linked chains [1] |
| Cell Assay |
Tumour cell-line panel viability. [1] GNE-6640 and GNE-6641 were profiled for 3 days across 441 cell lines, and GNE-6776, GNE-6640, and GNE-6641 were profiled for 5 days across a subset of 185 cell lines as previously described26. In brief, compounds were screened in nine-point dose–response using a threefold dilution. Cells were seeded into 384-well plates 24 h before compound addition. Cells were then incubated with compound for 72 h or 120 h before assaying viability. Assays were performed in biological triplicate. Cells were incubated (37 °C, 5% CO2) in RPMI-1640, 2.5% FBS (72 h assay) or 5% FBS (120 h assay), and 2 mM glutamine throughout the assay. The reported IC50 and mean viability metrics were as follows: IC50 was the dose at which the estimated inhibition was 50% relative to untreated wells (that is, absolute IC50). Primary combination screen. [1] A compound library comprising 589 compounds arrayed in nine-point dose–response was screened in the absence or presence of fixed doses of GNE-6776 (0 nm, 125 nM, 250 nM, 500 nM, 1,000 nM, and 2,000 nM) or GNE-6640 (400 nM). In brief, 5,000 EOL-1 cells were seeded into 384-well plates, and compound was added 24 h later. Cell viability was determined 120 h after compound addition (CellTiter-Glo). Curves were fitted, and both IC50 and mean viability metrics were calculated. The IC50 was the dose at which inhibition was 50% relative to untreated wells. The mean viability was the average of the fitted viabilities at each tested dose. Mean viability was equivalent to the area under the log-dose/viability curve divided by the total number of tested doses. Mean viability values were used for the analysis described in Extended Data Fig. 6g. All data were fitted using Genedata Screener software. Primary combination screen analysis. [1] Normalized mean viabilities were determined in the EOL-1 cell line for 574 compounds that have known protein or mechanistic targets, in the presence of DMSO or increasing concentrations of GNE-6776 (100 nM, 250 nM, 500 nM, 1,000 nM or 2,000 nM) or 400 nM of GNE-6640. For each compound, we assessed the difference in mean viability between USP7 inhibitor treatment and the DMSO treatment. For targets targeted by three or more compounds, we calculated the enrichment of high mean viability difference for each concentration of USP7 inhibitor by using a Wilcoxon rank-sum test. For visualization purposes, we combined the results of all concentrations by taking the mean of the −log10(transformed P values) for each target. 1. Tumor cell viability assay: Tumor cell lines were cultured in appropriate medium under standard cell culture conditions (e.g., 37°C, 5% CO₂). The cells were seeded into multi-well plates at a suitable density and allowed to adhere. Then, GNE-6776 at different concentrations was added to the wells, and the cells were incubated for a specific period (e.g., 24–72 hours). In combination experiments, chemotherapeutic agents or PIM kinase inhibitors were added to the wells along with GNE-6776. After incubation, a cell viability detection reagent (e.g., MTT or CCK-8) was added, and the absorbance was measured using a microplate reader to calculate cell viability. The assay demonstrated that GNE-6776 induces tumor cell death and enhances the cytotoxicity of other agents [1] 2. Western blot analysis for USP7 substrate proteins: Tumor cells were treated with GNE-6776 at different concentrations for a specific time. After treatment, the cells were harvested and lysed using a cell lysis buffer containing protease inhibitors to extract total cellular proteins. The protein concentration was determined, and equal amounts of proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were transferred to a nitrocellulose or PVDF membrane, which was then blocked with a blocking buffer (e.g., 5% non-fat milk). The membrane was incubated with primary antibodies specific to USP7 substrate proteins (such as p53 or MDM2) and a loading control protein (such as β-actin) overnight at 4°C. After washing, the membrane was incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody. Finally, the protein bands were visualized using an enhanced chemiluminescence (ECL) detection system. The results could reflect changes in the expression levels of USP7 substrate proteins after GNE-6776 treatment, indirectly confirming the inhibition of USP7 activity [1] |
| Animal Protocol |
Immunodeficient C.B-17 SCID mice with an EOL1 AML xenograft, aged 12–16 weeks[1] 100 or 200 mg/kg DMPK analysis. [1] In vitro DMPK studies were performed using standard protocols. GNE-6776 was formulated as a suspension in 0.5% methylcellulose/0.2% Tween-80 and was administered at 200 mg kg−1 (body weight) by oral gavage to female C.B-17 SCID mice, aged 12–16 weeks (n = 3 per time point). No randomization was used for DMPK studies. At 0.5, 1, 2, 4, 8 and 24 h post-dose, blood samples were collected by terminal cardiac puncture into anticoagulant tubes (EDTA). Clarified plasma was then transferred to a fresh tube and snap frozen. GNE-6776 plasma concentrations were determined by LC–MS/MS. In vivo pharmacodynamic response. [1] For EOL-1 AML xenograft studies, immunodeficient C.B-17 SCID mice, aged 12–16 weeks, were inoculated subcutaneously on the right flank with five million cells in a 50:50 suspension of HBSS:Matrigel (100 μl). When tumour volumes reached between approximately 285 and 500 mm3, mice were distributed into volume-matched cohorts (n = 4). For MCF7 breast-cancer xenograft studies, immunodeficient nu/nu mice, aged 6–8 weeks, were implanted with 0.36 mg oestrogen pellets via trochar 1–3 days before tumour cell inoculation. Ten million MCF7-Ser cells, an in vivo-optimized MCF7 variant, were injected orthotopically into the 2/3 mammary fat pad of each mouse in a 50:50 suspension of HBSS:Matrigel in a total volume of 100 μl. When tumour volumes reached between approximately 285 and 450 mm3, mice were distributed into volume-matched cohorts (n = 4). GNE-6776 was formulated as a suspension in 0.5% methylcellulose/0.2% Tween-80 and administered at 200 mg kg−1 (body weight) by oral gavage at 0 and 4 h. 0.5% Methylcellulose/0.2% Tween-80 control or GNE-6776-treated samples were collected at 8 h after the first dose and excised tumours were flash-frozen on dry ice. Tumours were lysed in RIPA buffer containing protease inhibitors and 300 mM NaCl using a Qiagen TissueLyser. Samples were incubated on ice for 15 min and then centrifuged at 20,000g at 4 °C for 10 min. Protein levels in clarified lysates were quantified using a Pierce BCA assay kit and concentrations were normalized with sample buffer. Samples were run on gels, and proteins were transferred to membranes and western blotted as described above. In vivo efficacy study. [1] For EOL1 AML xenograft studies, immunodeficient C.B-17 SCID mice (Charles River Laboratories), aged 12–16 weeks, were inoculated subcutaneously on the right flank with five million cells in a 50:50 suspension of HBSS:Matrigel (100 μl). When tumours became established (150–300 mm3), mice were distributed into tumour-volume-matched cohorts (n = 7, mean tumour volume ~250 mm3). GNE-6776 was formulated as a suspension in 0.5% methylcellulose/0.2% Tween-80 and was administered at 100 or 200 mg kg−1 (body weight) by oral gavage on a once or twice daily schedule. Tumour volume measurements, body weight and body condition data were collected two or three times per week. The maximum tumour volume limit of 2,000 mm3 was not reached in any animal. |
| References |
[1]. USP7 Small-Molecule Inhibitors Interfere With Ubiquitin Binding. Nature. 2017 Oct 26;550(7677):534-538. |
| Additional Infomation |
The ubiquitin system regulates essential cellular processes in eukaryotes. Ubiquitin is ligated to substrate proteins as monomers or chains and the topology of ubiquitin modifications regulates substrate interactions with specific proteins. Thus ubiquitination directs a variety of substrate fates including proteasomal degradation. Deubiquitinase enzymes cleave ubiquitin from substrates and are implicated in disease; for example, ubiquitin-specific protease-7 (USP7) regulates stability of the p53 tumour suppressor and other proteins critical for tumour cell survival. However, developing selective deubiquitinase inhibitors has been challenging and no co-crystal structures have been solved with small-molecule inhibitors. Here, using nuclear magnetic resonance-based screening and structure-based design, we describe the development of selective USP7 inhibitors GNE-6640 and GNE-6776. These compounds induce tumour cell death and enhance cytotoxicity with chemotherapeutic agents and targeted compounds, including PIM kinase inhibitors. Structural studies reveal that GNE-6640 and GNE-6776 non-covalently target USP7 12 Å distant from the catalytic cysteine. The compounds attenuate ubiquitin binding and thus inhibit USP7 deubiquitinase activity. GNE-6640 and GNE-6776 interact with acidic residues that mediate hydrogen-bond interactions with the ubiquitin Lys48 side chain, suggesting that USP7 preferentially interacts with and cleaves ubiquitin moieties that have free Lys48 side chains. We investigated this idea by engineering di-ubiquitin chains containing differential proximal and distal isotopic labels and measuring USP7 binding by nuclear magnetic resonance. This preferential binding protracted the depolymerization kinetics of Lys48-linked ubiquitin chains relative to Lys63-linked chains. In summary, engineering compounds that inhibit USP7 activity by attenuating ubiquitin binding suggests opportunities for developing other deubiquitinase inhibitors and may be a strategy more broadly applicable to inhibiting proteins that require ubiquitin binding for full functional activity.[1] Herein we describe GNE-6640 and GNE-6776, selective USP7 inhibitors that possess a structurally defined mechanism of inhibition. Establishing stringent screening cascades was critical for selecting and optimizing on-target inhibitors. Combination studies revealed a previously undescribed intersection between USP7 deubiquitinase activity and PIM kinases in regulating cell viability. Co-crystal structures of GNE-6640 or GNE-6776 pointed to the importance of the complementary charged interactions between USP7-D305/E308 and ubiquitin-K48 side chains, which we confirmed using mutational analysis. Notably, D305G has been identified as a somatic loss-of-function mutant in patients with acute lymphoblastic leukaemia21. NMR analysis of USP7 binding to native mono-ubiquitin and differentially labelled di-ubiquitins revealed that USP7 preferentially interacts with ubiquitin moieties having free K48 side chains. It has been proposed that the inefficiency of some deubiquitinases to depolymerize longer substrate-conjugated K48-linked chains enables a threshold for proteasome-targeting polyubiquitination22; our studies substantiate this idea and provide a biophysical mechanism. Numerous proteins, including other deubiquitinases, ubiquitin ligases, DNA repair and endocytosis machinery, and epigenetic regulators are functionally dependent on ubiquitin binding23. Developing selective inhibitors that attenuate ubiquitin binding is an effective strategy for USP7 inhibition. Our studies demonstrate the feasibility of this approach, which may have broader applications for inhibiting other classes of ubiquitin-binding proteins. [1] 1. The ubiquitin system plays a crucial role in regulating essential cellular processes in eukaryotes. Ubiquitin is ligated to substrate proteins as monomers or chains, and the topology of ubiquitin modifications regulates substrate interactions with specific proteins, thereby directing various substrate fates including proteasomal degradation. Deubiquitinase enzymes, such as USP7, cleave ubiquitin from substrates and are implicated in diseases [1] 2. USP7 regulates the stability of the p53 tumor suppressor and other proteins critical for tumor cell survival, making it a potential therapeutic target for cancer. However, developing selective deubiquitinase inhibitors has been challenging, and no co-crystal structures of USP7 with small-molecule inhibitors had been solved before this study [1] 3. GNE-6776 was developed through nuclear magnetic resonance-based screening and structure-based design, along with another USP7 inhibitor GNE-6640. Both compounds are selective USP7 inhibitors that non-covalently target USP7. The development of GNE-6776 suggests opportunities for developing other deubiquitinase inhibitors and may be a strategy broadly applicable to inhibiting proteins that require ubiquitin binding for full functional activity [1] |
Solubility Data
| Solubility (In Vitro) |
DMSO : 70~100 mg/mL ( 200.91~287.03 mM ) Ethanol : ~2 mg/mL |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (7.18 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (7.18 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (7.18 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8703 mL | 14.3513 mL | 28.7026 mL | |
| 5 mM | 0.5741 mL | 2.8703 mL | 5.7405 mL | |
| 10 mM | 0.2870 mL | 1.4351 mL | 2.8703 mL |