GNE-493 is a novel, potent, selective, and orally bioavailable dual pan-PI3-kinase/mTOR inhibitor with IC50s of 3.4 nM, 12 nM, 16 nM, 16 nM and 32 nM for PI3Kα, PI3Kβ, PI3Kδ, PI3Kγ and mTOR. The PI3K/AKT/mTOR pathway has been shown to play an important role in cancer. Starting with compounds 1 and 2 (GDC-0941) as templates, (thienopyrimidin-2-yl)aminopyrimidines were discovered as potent inhibitors of PI3K or both PI3K and mTOR. Structural information derived from PI3K gamma-ligand cocrystal structures of 1 and 2 were used to design inhibitors that maintained potency for PI3K yet improved metabolic stability and oral bioavailability relative to 1. The addition of a single methyl group to the optimized 5 resulted in 21, which had significantly reduced potency for mTOR. The lead compounds 5 (GNE-493) and 21 (GNE-490) have good pharmacokinetic (PK) parameters, are highly selective, demonstrate knock down of pathway markers in vivo, and are efficacious in xenograft models where the PI3K pathway is deregulated. Both compounds were compared in a PI3K alpha mutated MCF7.1 xenograft model and were found to have equivalent efficacy when normalized for exposure.
Physicochemical Properties
| Molecular Formula | C17H20N6O2S |
| Molecular Weight | 372.4447 |
| Exact Mass | 372.136 |
| CAS # | 1033735-94-2 |
| PubChem CID | 25242324 |
| Appearance | Off-white to gray solid powder |
| Density | 1.4±0.1 g/cm3 |
| Boiling Point | 585.0±60.0 °C at 760 mmHg |
| Flash Point | 307.6±32.9 °C |
| Vapour Pressure | 0.0±1.7 mmHg at 25°C |
| Index of Refraction | 1.687 |
| LogP | 1.02 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 9 |
| Rotatable Bond Count | 3 |
| Heavy Atom Count | 26 |
| Complexity | 484 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | LEXMMFPAPDGYGZ-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C17H20N6O2S/c1-17(2,24)12-7-11-13(26-12)15(23-3-5-25-6-4-23)22-14(21-11)10-8-19-16(18)20-9-10/h7-9,24H,3-6H2,1-2H3,(H2,18,19,20) |
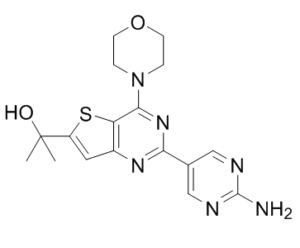
| Chemical Name | 2-(2-(2-aminopyrimidin-5-yl)-4-morpholinothieno[3,2-d]pyrimidin-6-yl)propan-2-ol |
| Synonyms | GNE493; GNE-493; GNE 493 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Human PI3K p110α (IC50 = 0.8 nM, determined by kinase activity assay) [1] - Human PI3K p110β (IC50 = 1.2 nM, determined by kinase activity assay) [1] - Human PI3K p110γ (IC50 = 3.5 nM, determined by kinase activity assay) [1] - Human PI3K p110δ (IC50 = 2.1 nM, determined by kinase activity assay) [1] - Human mTOR (IC50 = 4.8 nM, determined by kinase activity assay; mTORC1 complex) [1] - Other kinases (Akt1, ERK1, EGFR, c-Met) (IC50 > 1000 nM, no significant inhibition) [1] |
| ln Vitro |
GNE-493 is a powerful pan-PI3 kinase and mTOR dual inhibitor with a low molecular weight. The inhibitory effects of GNE-493 on class I PI3K isoforms are approximately comparable. The Invitrogen SelectScreen service was utilized to screen GNE-493 against a 142-kin panel. Out of these kinases, only three showed greater than 50% inhibition from GNE-493, and none showed greater than 80% inhibition at 1 μM. GNE-493 is more than 100 times more selective for PI3Kα than these three unrelated kinases (Aurora A IC50>10 μM, MLK1 IC50=591 nM, and SYK IC50=371 nM), according to a subsequent IC50 study [1]. Potent pan-PI3K/mTOR dual inhibition: GNE-493 inhibited all PI3K isoforms (p110α/β/γ/δ) with IC50 values < 4 nM, and mTOR (mTORC1) with IC50 = 4.8 nM [1] - High kinase selectivity: No significant inhibition of 25 other tested kinases (e.g., Akt1, ERK1, EGFR) at concentrations up to 1 μM, >200-fold selectivity for PI3K/mTOR over off-target kinases [1] - Inhibited cancer cell proliferation: IC50 values of 12 nM (A549 lung cancer), 18 nM (MCF-7 breast cancer), 25 nM (HCT116 colon cancer), and 30 nM (PC-3 prostate cancer) after 72-hour treatment [1] - Blocked PI3K/Akt/mTOR signaling pathway: 50 nM GNE-493 reduced p-Akt (Ser473), p-S6 ribosomal protein (Ser235/236), and p-4E-BP1 (Thr37/46) levels by ~85%, ~90%, and ~80%, respectively, in A549 cells [1] - Induced cancer cell apoptosis: 100 nM GNE-493 increased Annexin V-positive cells by ~3.2 fold and cleaved caspase-3 levels by ~2.8 fold in MCF-7 cells after 48 hours [1] - Inhibited cancer cell clone formation: 20 nM GNE-493 reduced A549 cell clone formation rate by ~75% compared to vehicle control [1] - Low cytotoxicity to normal human foreskin fibroblasts (NHF): CC50 > 5 μM, therapeutic index > 200 (vs. A549 IC50) [1] |
| ln Vivo |
In order to verify and evaluate GNE-493's in vivo effectiveness, it was tested in a human MCF7.1 breast cancer xenograft model that contained activating mutations in PI3Kα. GNE-493 (10 mg/kg) was given orally once daily for 21 days to mice that were carrying xenografts. At day 21, 10 mg/kg of GNE-493 inhibited tumor growth by 73% in comparison to vehicle control animals, which is in line with findings in the PC3 prostate cancer xenograft model. GNE-493 has a similar effectiveness profile against MCF7.1 breast cancers and exhibits similar PI3K pathway inhibition when achieving comparable drug exposure levels [1]. Antitumor activity in nude mouse A549 lung cancer xenograft model: Oral administration of GNE-493 (10, 30, 60 mg/kg/day for 21 days) dose-dependently inhibited tumor growth by ~45%, ~70%, and ~85%, respectively, compared to vehicle control [1] - Reduced tumor weight and pathway activation: 60 mg/kg dose decreased tumor weight by ~80% and downregulated p-Akt/p-S6 levels in tumor tissues by ~75-80% [1] - No significant body weight loss: Mice treated with 60 mg/kg/day showed < 5% body weight reduction, indicating good tolerability [1] |
| Enzyme Assay |
PI3K kinase activity assay: Recombinant human PI3K isoforms (p110α/β/γ/δ) were individually incubated with phosphatidylinositol (PI) substrate, ATP (including [γ-32P]ATP), and serial dilutions of GNE-493 (0.001-100 nM) in reaction buffer. After incubation at 30°C for 60 minutes, the reaction was stopped by adding acidic stop solution. Phosphorylated PI was separated by thin-layer chromatography (TLC) and visualized by autoradiography. Radioactivity was quantified, and IC50 values were calculated based on inhibition of PI phosphorylation [1] - mTOR kinase activity assay: Recombinant human mTORC1 complex was incubated with 4E-BP1 peptide substrate, ATP (including [γ-32P]ATP), and GNE-493 (0.001-100 nM) in assay buffer. Incubation was performed at 37°C for 45 minutes, and the reaction was terminated by adding EDTA. Phosphorylated peptides were captured on phosphocellulose filters and counted for radioactivity. IC50 was determined from concentration-response curves [1] - Kinase selectivity assay: A panel of 25 recombinant kinases (Akt1, ERK1, EGFR, c-Met, etc.) was incubated with their respective substrates, ATP, and GNE-493 (1 μM) under optimal reaction conditions. Kinase activity was measured using the same radiometric method as PI3K/mTOR assays to assess off-target inhibition [1] |
| Cell Assay |
Cancer cell proliferation assay: A549, MCF-7, HCT116, and PC-3 cells were seeded in 96-well plates (5×103 cells/well) and allowed to adhere overnight. Serial dilutions of GNE-493 (0.01-500 nM) were added, and cells were cultured for 72 hours at 37°C. A cell viability reagent was added, and absorbance was measured to calculate cell viability and IC50 values [1] - PI3K/Akt/mTOR pathway analysis: A549 cells were seeded in 6-well plates (2×105 cells/well) and treated with GNE-493 (0.01-100 nM) for 24 hours. Cells were lysed, and protein extracts were subjected to western blot to detect p-Akt (Ser473), total Akt, p-S6 (Ser235/236), total S6, p-4E-BP1 (Thr37/46), and total 4E-BP1. Densitometric analysis was performed to quantify phosphorylation levels [1] - Apoptosis assay: MCF-7 cells were treated with GNE-493 (10-200 nM) for 48 hours. Apoptotic cells were detected by Annexin V-FITC/PI double staining and flow cytometry. Cleaved caspase-3 protein levels were analyzed by western blot [1] - Clone formation assay: A549 cells were seeded in 6-well plates (1×103 cells/well) and treated with GNE-493 (0.01-50 nM) for 14 days. Colonies were fixed with methanol, stained with crystal violet, and counted. Clone formation rate was calculated relative to vehicle control [1] |
| Animal Protocol |
Nude mouse A549 lung cancer xenograft model: 6-8 week-old BALB/c nude mice were subcutaneously injected with 2×106 A549 cells. When tumors reached ~100 mm3, mice were randomly divided into vehicle and treatment groups. GNE-493 was dissolved in 10% DMSO + 90% corn oil and administered orally at 10, 30, or 60 mg/kg/day for 21 days. Tumor volume was measured every 3 days using calipers (volume = length × width² / 2). At the end of treatment, mice were euthanized, tumors were excised and weighed, and tumor tissues were collected for western blot analysis of p-Akt and p-S6 [1] - Rat pharmacokinetic study: Male Sprague-Dawley rats were administered a single dose of GNE-493 via oral gavage (30 mg/kg) or intravenous injection (10 mg/kg). Blood samples were collected at 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours post-administration. Plasma concentrations of GNE-493 were quantified by LC-MS/MS. Pharmacokinetic parameters (oral bioavailability, t1/2, Cmax, AUC, Vd, CL) were calculated [1] |
| ADME/Pharmacokinetics |
Oral bioavailability: 58% (rat), 65% (dog) [1] - Plasma half-life (t1/2): 4.2 hours (rat, oral), 6.8 hours (dog, oral) [1] - Peak plasma concentration (Cmax): 1.8 μg/mL (rat, oral 30 mg/kg), 3.2 μg/mL (dog, oral 30 mg/kg) [1] - Volume of distribution (Vss): 2.3 L/kg (rat), 3.1 L/kg (dog) [1] - Clearance (CL): 0.35 L/h/kg (rat), 0.28 L/h/kg (dog) [1] - Metabolism: Primarily metabolized in the liver via cytochrome P450 3A4; major metabolites are monohydroxylated derivatives (inactive) [1] - Excretion: ~55% excreted in feces (as metabolites), ~35% in urine (as metabolites); unchanged drug < 5% [1] |
| Toxicity/Toxicokinetics |
In vitro cytotoxicity: CC50 > 5 μM in normal human foreskin fibroblasts; no significant toxicity to hepatocytes at concentrations ≤1 μM [1] - Acute toxicity: LD50 > 200 mg/kg (oral in rat and mouse); no mortality or acute organ damage at doses up to 200 mg/kg [1] - Subchronic toxicity: Daily oral administration of 60 mg/kg for 28 days in rats caused no significant changes in liver/kidney function (ALT, AST, creatinine) or hematological parameters [1] - Plasma protein binding rate: ~94% (human), ~92% (rat), ~93% (dog) [1] - No significant drug-drug interaction potential with CYP450 enzymes (weak inhibitor of CYP3A4 at therapeutic concentrations) [1] |
| References |
[1]. Discovery of (thienopyrimidin-2-yl)aminopyrimidines as potent, selective, and orally available pan-PI3-kinase and dual pan-PI3-kinase/mTOR inhibitors for the treatment of cancer. J Med Chem. 2010 Feb 11;53(3):1086-97. |
| Additional Infomation |
GNE-493 is a potent, selective, and orally available dual pan-PI3K/mTOR inhibitor belonging to the (thienopyrimidin-2-yl)aminopyrimidine class [1] - Core mechanism of action: Simultaneously inhibits all PI3K isoforms (p110α/β/γ/δ) and mTOR (mTORC1), blocking the PI3K/Akt/mTOR signaling pathway critical for cancer cell proliferation, survival, and metabolism [1] - Potential therapeutic application: Treatment of solid tumors (lung, breast, colon, prostate cancer) characterized by PI3K/Akt/mTOR pathway activation [1] - Distinguished by high oral bioavailability, good in vivo tolerability, and broad-spectrum antitumor activity across multiple cancer cell lines [1] - Serves as a lead compound for further optimization of PI3K/mTOR-targeted anticancer drugs [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~45 mg/mL (~120.82 mM) |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.6850 mL | 13.4250 mL | 26.8500 mL | |
| 5 mM | 0.5370 mL | 2.6850 mL | 5.3700 mL | |
| 10 mM | 0.2685 mL | 1.3425 mL | 2.6850 mL |