Physicochemical Properties
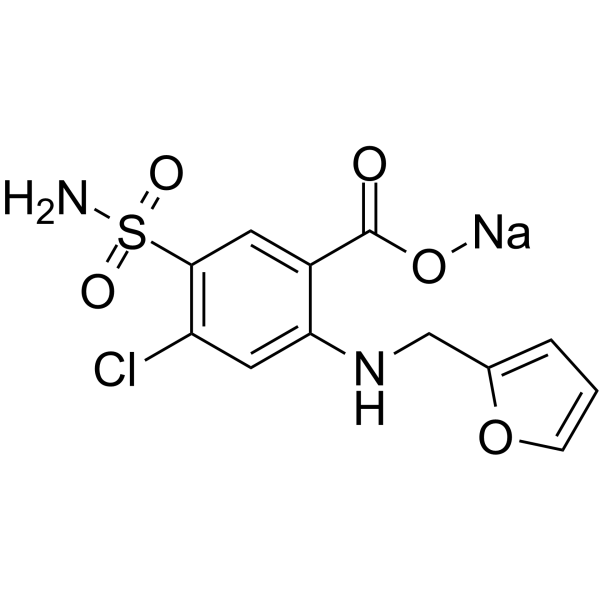
| Molecular Formula | C12H10N2O5SCL-.NA+ |
| Molecular Weight | 352.726 |
| Exact Mass | 351.99 |
| Elemental Analysis | C, 40.86; H, 2.86; Cl, 10.05; N, 7.94; Na, 6.52; O, 22.68; S, 9.09 |
| CAS # | 41733-55-5 |
| Related CAS # | Furosemide;54-31-9; Furosemide-d5;1189482-35-6;42461-27-8 (HCl); 54-31-9; 41733-55-5 (sodium); 61422-49-9 (xantinol) |
| PubChem CID | 23673593 |
| Appearance | White to off-white solid powder |
| Boiling Point | 582.1ºC at 760 mmHg |
| Melting Point | 206ºC |
| Flash Point | 305.9ºC |
| LogP | 2.41 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 5 |
| Heavy Atom Count | 22 |
| Complexity | 486 |
| Defined Atom Stereocenter Count | 0 |
| InChi Key | DLFCAVBMDSKMEY-UHFFFAOYSA-M |
| InChi Code | InChI=1S/C12H11ClN2O5S.Na/c13-9-5-10(15-6-7-2-1-3-20-7)8(12(16)17)4-11(9)21(14,18)19;/h1-5,15H,6H2,(H,16,17)(H2,14,18,19);/q;+1/p-1 |
| Chemical Name | sodium;4-chloro-2-(furan-2-ylmethylamino)-5-sulfamoylbenzoate |
| Synonyms | Sodium furosemide; Furosemide sodium; 41733-55-5; Furosemide (sodium); Benzoic acid, 5-(aminosulfonyl)-4-chloro-2-[(2-furanylmethyl)amino]-,monosodium salt; 101EM454S7; sodium;4-chloro-2-(furan-2-ylmethylamino)-5-sulfamoylbenzoate; Frosemide sodium; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Na+/2Cl-/K+ (NKCC) symporter; NKCC1/2; GABAA receptors - Furosemide sodium targets K-Cl cotransporter (KCC) in rabbit, rat, and human tissues[1] - Furosemide sodium targets gamma-aminobutyric acid type AA (GABAAA) receptor subtypes (selective antagonism on specific subtypes)[2] - Furosemide sodium targets Na + /K + /2Cl - cotransporter (NKCC) in human gastric cancer cells [4] - Furosemide sodium targets non-selective voltage-independent cation channels (NS-VICC) in human erythrocytes (IC50 = 100 μM for channel current inhibition)[5] |
| ln Vitro |
MKN45 cells (hypouric human adenoid lineage) exhibit a considerable change in proliferation rate when exposed to furosemide (500 μM; 72-96 hours). On MKN28 cells, however, it exhibited little effect (moderate urothelial adenoids). When exposed to furosemide sodium (10 μM, 30 μM, or 100 μM for 45 minutes), MKN45 cells grow at the fastest rate compared to MKN28 cells [4]. considerably lowers the activity of cartilage channels[5]. - Inhibition of K-Cl cotransporter activity: In Xenopus oocytes expressing recombinant rabbit, rat, or human KCC, Furosemide sodium (100 μM) inhibited K + transport (measured via 86 Rb + uptake) by 65%–72% compared to the non-inhibited control. The inhibition was concentration-dependent, with 50 μM causing 30%–35% inhibition[1] - Antagonism of GABAAA receptor subtypes: In HEK293 cells expressing recombinant GABAAA receptor subtypes (α1β2γ2, α6β2γ2), Furosemide sodium (1 mM) selectively inhibited GABA-induced chloride currents in α6β2γ2 subtype by 58%, while having no significant effect on α1β2γ2 subtype (inhibition < 10%). The antagonism was reversible after drug washout[2] - Inhibition of human gastric cancer cell proliferation: In poorly differentiated human gastric cancer cells (MKN-45), Furosemide sodium (50, 100, 200 μM) inhibited proliferation in a concentration-dependent manner. After 72-hour incubation, 200 μM reduced cell viability by 48% (MTT assay) and increased the proportion of cells in G0/G1 phase from 45% (control) to 68% (flow cytometry). It had no significant effect on normal human gastric epithelial cells at 200 μM[4] - Inhibition of NS-VICC in human erythrocytes: In patch-clamp experiments on human erythrocyte membranes, Furosemide sodium inhibited NS-VICC-mediated cation currents. At 100 μM (IC50), the current amplitude was reduced by 50%; at 200 μM, inhibition reached 85%. The inhibition was not affected by voltage (proving voltage independence of the channel)[5] |
| ln Vivo |
C57BL/6 mice were utilized to generate a deaf-mute model with kanamycin (KM) (1000 mg/kg) and furosemide sodium injection (ip; 100 mg/kg; single dose). On days 1, 2, and 3, following injection, hearing impairment and cochlear hair cell destruction were measured, accordingly. Day 3 of the mice's OHC (outer hair cell) morphology of the top circle, middle circle, and eyeball circle revealed that hearing was clearly impaired even from the second day (day 1 group) [1]. - Ototoxicity in kanamycin/furosemide-treated mice: C57BL/6 mice were given a single intraperitoneal injection of kanamycin (400 mg/kg) followed by Furosemide sodium (400 mg/kg, intraperitoneal) 30 minutes later. This combination induced severe hearing loss: auditory brainstem response (ABR) thresholds at 8 kHz, 16 kHz, and 32 kHz increased by 45 dB, 52 dB, and 60 dB, respectively, compared to the control group (saline-injected). Pretreatment with peptide vaccine GV1001 (100 μg/mouse, subcutaneous, 3 times at 7-day intervals) rescued hearing, reducing ABR threshold increases by 30%–35%[3] |
| Enzyme Assay |
GABAA receptors in cerebellar granule cells are unique in expressing a subtype containing the alpha6 subunit. This receptor subtype has high affinity for GABA and produces a degree of tonic inhibition on cerebellar granule cells, modulating the firing of these cells via spillover of GABA from GABAergic synapses. This receptor subtype also has selective affinity for the diuretic furosemide over receptors containing other alpha-subunits. Furosemide exhibits approximately 100-fold selectivity for alpha6-containing receptors over alpha1-containing receptors. By making alpha1/alpha6 chimeras we have identified a transmembrane region (209-279) responsible for the high furosemide sensitivity of alpha6beta3gamma2s receptors. Within the alpha1 transmembrane region, a single amino acid was identified that when mutated from threonine to isoleucine, increased furosemide sensitivity by 20-fold. We demonstrate the beta-subunit selectivity of furosemide to be due to asparagine 265 in the beta2 and beta3 transmembrane-domain II similar to that observed with potentiation by the anticonvulsant loreclezole. We also show that Ile in transmembrane-domain I accounts for the increased GABA sensitivity observed at alpha6beta3gamma2s compared with alpha1beta3gamma2s receptors, but did not affect direct activation by pentobarbital or potentiation by the benzodiazepine flunitrazepam. Location of these residues within transmembrane domains leads to speculation that they may be involved in the channel-gating mechanism conferring increased receptor activation by GABA, in addition to conferring furosemide sensitivity.[2] - K-Cl cotransporter activity assay (via 86 Rb + uptake): Xenopus oocytes were injected with KCC cRNA (rabbit, rat, or human) and cultured for 48 hours. Oocytes were incubated in buffer containing 86 RbCl (1 μCi/mL) and different concentrations of Furosemide sodium (25, 50, 100 μM) for 1 hour. After washing to remove unincorporated 86 Rb + , the radioactivity in oocytes was measured using a gamma counter. KCC activity was calculated as the difference in 86 Rb + uptake between KCC-expressing and non-expressing oocytes[1] - NS-VICC current recording (patch-clamp technique): Human erythrocytes were isolated and attached to a coverslip in a recording chamber. The whole-cell patch-clamp configuration was established using a glass pipette filled with intracellular solution. Furosemide sodium (50, 100, 200 μM) was added to the extracellular solution sequentially. Cation currents were recorded at holding potentials of -60 mV, -40 mV, 0 mV, +40 mV, and +60 mV. Current amplitude was analyzed using electrophysiology software to calculate inhibition rate[5] |
| Cell Assay |
Furosemide, a blocker of Na(+)/K(+)/2Cl(-) cotransporter (NKCC), is often used as a diuretic to improve edema, ascites, and pleural effusion of patients with cancers. The aim of the present study was to investigate whether an NKCC blocker affects cancer cell growth. If so, we would clarify the mechanism of this action. We found that poorly differentiated gastric adenocarcinoma cells (MKN45) expressed the mRNA of NKCC1 three times higher than moderately differentiated ones (MKN28) and that the NKCC in MKN45 showed higher activity than that in MKN28. A cell proliferation assay indicates that furosemide significantly inhibited cell growth in MKN45 cells, but not in MKN28 cells. Using flow cytometrical analysis, we found that the exposure to furosemide brought MKN45 cells to spend more time at the G(0)/G(1) phase, but not MKN28 cells. Based on these observations, we indicate that furosemide diminishes cell growth by delaying the G(1)-S phase progression in poorly differentiated gastric adenocarcinoma cells, which show high expression and activity of NKCC, but not in moderately differentiated gastric adenocarcinoma cells with low expression and NKCC activity.[1] Background: Furosemide, a loop diuretic inhibiting the renal tubular Na(+),K(+),2Cl(-) cotransporter, has been shown to decrease cytosolic Ca(2+) concentration ([Ca(2+)](i)) in platelets and erythrocytes. [Ca(2+)](i) in erythrocytes is a function of Ca(2+) permeable cation channels. Activation of those channels e.g. by energy depletion or oxidative stress leads to increase of [Ca(2+)](i), which in turn triggers eryptosis, a suicidal erythrocyte death characterized by cell membrane scrambling. The present study was performed to explore whether furosemide influences the cation channels and thus influences eryptosis.[5] Methods: Cation channel activity was determined by whole-cell patch clamp, [Ca(2+)](i) utilizing Fluo3 fluorescence and annexin V binding to estimate cell membrane scrambling with phosphatidylserine exposure. [5] Results: A 45 min exposure to furosemide (10 and 100 µM) slightly, but significantly decreased cation channel activity and [Ca(2+)](i) in human erythrocytes drawn from healthy individuals. ATP-depletion (> 3 hours, +37°C, 6 mM ionosine and 6 mM iodoacetic acid) enhanced the non-selective cation channel activity, increased [Ca(2+)](i) and triggered cell membrane scrambling, effects significantly blunted by furosemide (10 - 100 µM). Oxidative stress by exposure to tert-butylhydroperoxide (0.1 -1 mM) similarly enhanced the non-selective cation channels activity, increased [Ca(2+)](i) and triggered cell membrane scrambling, effects again significantly blunted by furosemide (10 - 100 µM).[5] Conclusions: The present study shows for the first time that the loop diuretic furosemide applied at micromolar concentrations (10 - 100 µM) inhibits non-selective cation channel activity in and eryptosis of human erythrocytes.[5] - Gastric cancer cell proliferation and cell cycle assay: Poorly differentiated human gastric cancer cells (MKN-45) were seeded into 96-well plates (for MTT) or 6-well plates (for flow cytometry) at 5×10³ cells/well or 2×10⁵ cells/well, respectively. After 24-hour adherence, Furosemide sodium (50, 100, 200 μM) was added. For MTT assay: cells were incubated for 72 hours, MTT reagent was added, and absorbance was measured at 570 nm to calculate viability. For flow cytometry: cells were harvested after 48-hour incubation, fixed with ethanol, stained with propidium iodide, and analyzed for cell cycle distribution (G0/G1, S, G2/M phases)[4] - GABAAA receptor current recording: HEK293 cells were transfected with GABAAA receptor subunit cDNAs (α1β2γ2 or α6β2γ2) and cultured for 48 hours. Whole-cell patch-clamp was used to record GABA-induced Cl - currents. GABA (10 μM) was applied to induce currents, then Furosemide sodium (100 μM, 500 μM, 1 mM) was co-applied. Current amplitude changes were recorded to assess antagonism[2] - Erythrocyte NS-VICC assay: Human erythrocytes were isolated from fresh blood via centrifugation and resuspended in physiological saline. Furosemide sodium (50, 100, 200 μM) was added, and the suspension was incubated at 37°C for 1 hour. Erythrocyte membrane potential was measured using a potentiometer, and cation influx (Na + , K + ) was quantified via atomic absorption spectroscopy. At 100 μM, cation influx was reduced by 50% compared to control[5] |
| Animal Protocol |
Deaf Mouse Model and Study Groups[3] A deaf mouse model (C57BL/6 mouse, 4–6 weeks of age, weight of 15–25 g) was created by intraperitoneal injection of KM (1000 mg/kg) followed by furosemide (100 mg/kg) within 30 min. In Experiment 1, to assess the initial temporal change of hearing and the extent of hair cell damage in this deaf mouse model, total nine mice were divided into three groups: Day-1 (N = 3), Day-2 (N = 3) and Day-3 (N = 3). After injection of KM and furosemide on day 0, hearing loss and cochlear hair cell damage were evaluated on day 1, day 2 and day 3, respectively (Supplementary file S1).[3] In Experiment 2, to test the rescue effect of GV1001, total 120 mice were divided into the following three treatment groups: GV1001 (N = 40), dexamethasone (N = 40) and saline (N = 40). GV1001 (10 mg/kg), dexamethasone (15 mg/kg), or saline was subcutaneously administered for three consecutive days after the injection of KM and furosemide. To compare the rescue effect of GV1001 on different time points, each group was divided into four subgroups according to the time points of GV1001, dexamethasone, and saline treatment: D0 group (days 0, 1 and 2), D1 group (days 1, 2 and 3), D3 group (days 3, 4 and 5), and D7 group (days 7, 8 and 9; Supplementary file S2).[3] A deaf mouse model was created by intraperitoneal injection of KM and furosemide. First, to test the early temporal change of hearing and extent of hair cell damage after KM and furosemide injection, hearing and outer hair cells (OHCs) morphology were evaluated on day 1, day 2 and day 3 after injection. In the second experiment, following KM and furosemide injection, GV1001, dexamethasone, or saline were given for three consecutive days at different time points: D0 group (days 0, 1, and 2), D1 group (days 1, 2, and 3), D3 group (days 3, 4, and 5) and D7 group (days 7, 8, and 9). The hearing thresholds were measured at 8, 16, and 32 kHz before ototoxic insult, and 7 days and 14 days after KM and furosemide injection. After 14 days, each turn of the cochlea was imaged to evaluate OHCs damage. GV1001-treated mice showed significantly less hearing loss and OHCs damage than the saline control group in the D0, D1 and D3 groups (p < 0.0167). However, there was no hearing restoration or intact hair cell in the D7 group. GV1001 protected against cochlear hair cell damage, and furthermore, delayed administration of GV1001 up to 3 days rescued hair cell damage and hearing loss in KM/furosemide-induced deaf mouse model.[3] - Mouse kanamycin/Furosemide sodium-induced ototoxicity model: Female C57BL/6 mice (8–10 weeks old) were divided into 3 groups (n=8/group): 1) Control group: intraperitoneal injection of saline; 2) Ototoxicity group: intraperitoneal injection of kanamycin (400 mg/kg) followed by Furosemide sodium (400 mg/kg, intraperitoneal) 30 minutes later; 3) GV1001 pretreatment group: subcutaneous injection of GV1001 (100 μg/mouse) on days 0, 7, and 14, then treated with kanamycin/Furosemide sodium on day 21. On day 28, ABR was measured to assess hearing function (stimuli: 8 kHz, 16 kHz, 32 kHz tone bursts). After ABR testing, mice were sacrificed, and cochleae were collected for histological analysis[3] |
| ADME/Pharmacokinetics |
Absorption Following oral administration, furosemide is absorbed from the gastrointestinal tract. It displays variable bioavailability from oral dosage forms, ranging from 10 to 90%. The oral bioavailability of furosemide from oral tablets or oral solution is about 64% and 60%, respectively, of that from an intravenous injection of the drug. Route of Elimination The kidneys are responsible for 85% of total furosemide total clearance, where about 43% of the drug undergoes renal excretion. Significantly more furosemide is excreted in urine following the I.V. injection than after the tablet or oral solution. Approximately 50% of the furosemide load is excreted unchanged in urine, and the rest is metabolized into glucuronide in the kidney. Volume of Distribution The volume of distribution following intravenous administration of 40 mg furosemide were 0.181 L/kg in healthy subjects and 0.140 L/kg in patients with heart failure. Clearance Following intravenous administration of 400 mg furosemide, the plasma clearance was 1.23 mL/kg/min in patients with heart failure and 2.34 mL/kg/min in healthy subjects, respectively. Significantly more furosemide is excreted in urine following the IV injection than after the tablet or oral solution. There are no significant differences between the two oral formulations in the amount of unchanged drug excreted in urine. NIH; DailyMed. Current Medication Information for Lasix (Furosemide Tablet) (Updated: April 2016). Available from, as of April 19, 2017: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=2c9b4d8f-0770-482d-a9e6-9c616a440b1a After oral administration of furosemide to 18 pregnant women on the day of delivery, substantial concentrations of the drug were detected in umbilical cord vein plasma as well as in amniotic fluid. The ratio between the furosemide concentrations in maternal vein plasma and in umbilical cord plasma increased with time and approximated unity at 8 to 10 hr after administration of the drug. The plasma half-life of furosemide appeared to be longer in the mothers than in nonpregnant healthy volunteers. In one patient the plasma level of furosemide was constant during 5 hr of observation. PMID:699480 In one study in patients with normal renal function, approx 60% of a single 80 mg oral dose of furosemide was absorbed from the GI tract. When admin to fasting adults in this dosage, the drug appeared in the serum within 10 min, reached a peak concn of 2.3 ug/mL in 60-70 min, & was almost completely cleared from the serum in 4 hr. When the same dose was given after a meal, the serum concn of furosemide increased slowly to a peak of about 1 ug/ml after 2 hr & similar concns were present 4 hr after ingestion. However, a similar diuretic response occurred regardless of whether the drug was given with food or to fasting patients. In another study, the rate & extent of absorption varied considerably when 1 g of furosemide was given orally to uremic patients. An avg of 76% of a dose was absorbed, & peak plasma concns were achieved within 2-9 hr (avg 4.4 hr). Serum concns required to produce max diuresis are not known, & it has been reported that the magnitude of response does not correlate with either the peak or the mean serum concns. The diuretic effect of orally administered furosemide is apparent within 30 minutes to 1 hr and is maximal in the first or second hour. The duration of action is usually 6-8 hr. The maximum hypotensive effect may not be apparent until several days after furosemide therapy is begun. After iv administration of furosemide, diuresis occurs within 5 min, reaches a maximum within 20-60 min, and persists for approximately 2 hr. After im administration, peak plasma concentrations are attained within 30 min; onset of diuresis occurs somewhat later than after iv administration. In patients with severely impaired renal function, the diuretic response may be prolonged. View MoreMetabolism / MetabolitesThe metabolism of furosemide occurs mainly in the kidneys and the liver, to a smaller extent. The kidneys are responsible for about 85% of total furosemide total clearance, where about 40% involves biotransformation. Two major metabolites of furosemide are furosemide glucuronide, which is pharmacologically active, and saluamine (CSA) or 4-chloro-5-sulfamoylanthranilic acid. It would appear that frusemide glucuronide is the only or at least the major biotransformation metabolite in man. 2-amino-4- chloro-5-sulfamoylanthranilic acid has been reported in some studies but not in others; and is thought to be an analytical artifact. In patients with normal renal function, a small amount of furosemide is metabolized in the liver to the defurfurylated derivative, 4-chloro-5-sulfamoylanthranilic acid. ... American Society of Health-System Pharmacists 2016; Drug Information 2016. Bethesda, MD. 2016, p. 2831 Biological Half-Life The half-life from the dose of 40 mg furosemide was 4 hours following oral administration and 4.5 hours following intravenous administration. The terminal half-life of furosemide is approximately 2 hours following parenteral administration. The terminal half-life may be increased up to 24 hours in patients with severe renal failure. To study the pharmacokinetics of furosemide (fursemide; Lasix) and its acyl glucuronide and to analyze the pharmacodynamic response, a study was conducted in 7 healthy subjects, mean age 34 yr, who received a single oral 80 mg dose of furosemide in tablet form. Two half-lives were distinguished in the plasma elimination of furosemide and its conjugate, with values of 1.25 and 30.4 hr for furosemide and 1.31 and 33.2 hr for the conjugate. ... In dogs, ... the elimination half life /is/ approximately 1-1.5 hours. Various investigators have reported a wide range of elimination half-lives for furosemide. In one study, the elimination half-life averaged about 30 minutes in healthy patients who received 20-120 mg of the drug IV. In another study, the elimination half-life averaged 9.7 hours in patients with advanced renal failure who received 1 g of furosemide IV. The elimination half-life was more prolonged in 1 patient with concomitant liver disease. The serum half-life in therapeutic doses is 92 minutes; increasing in patients with uremia; congestive heart failure and cirrhosis as well as in the neonate and aged patients. In such patients the half-life may be extended to 20 hours. |
| Toxicity/Toxicokinetics |
Toxicity Summary IDENTIFICATION AND USE: Furosemide is white or slightly yellow, solid powder or crystals. Furosemide is used to treat edema associated with many diseases (either as the sole drug or as an adjunct to other antihypertensives). HUMAN STUDIES: Overdoses of diuretics are uncommon and infrequently serious. Problems most frequently involve chronic over medication or poor monitoring and/or lack of anticipation of drug interactions or not compensating for concomitant hepatic or renal dysfunction. Main toxic effects are on the kidneys with diuresis of water, sodium, and potassium leading most frequently to a hyponatremic, hypokalemic, and hypochloremic dehydration. More caution is warranted with patients at higher risk for abnormal renal function including patients with any renal disease, diabetes mellitis, and borderline fluid and/or electrolyte status. In a hospital-based study of adverse reactions to medications there was a 21% rate of adverse affects to frusemide with most common ones being hypovolemia, hyperuricemia, and hypokalemia which for the most part were mild, but the rate and severity increased with increasing daily doses. Hypersensitivity reactions such as rash, photosensitivity, thrombocytopenia, and pancreatitis are rare. Life threatening hyperkalemia and reversible renal failure occurred in an elderly male taking captopril concomitantly with furosemide. Acute rhabdomyolysis and myoglobinuria due to hypokalaemia occurred in a 74-year-old male taking furosemide. Severe anaphylactic reaction to furosemide involving urticaria; angioedema and hypotension occurred in an adult 5 minutes after receiving intravenous furosemide. Furosemide abuse (400 mg daily) was associated with severe hyponatremia and central pontine myelinolysis. Self-administration of furosemide over 6 years resulted in calcification of the renal medulla. Nephrocalcinosis and nephrolithiasis occurred in 5 children after treatment with furosemide. It has also reported nephrocalcinosis in premature infants being treated with furosemide. Cholelithiasis in infants was caused by furosemide. Tachycardia was reported following a high dose intravenous regimen using furosemide. Renal calcifications were encountered in premature infants when given furosemide. Furosemide-induced renal calcifications in low birth weight infants may lead to glomerular and tubular dysfunction in the long-term. A concentration-dependent increase in the frequency of chromosomal aberrations was observed in human lymphocytes exposed in vitro to furosemide for 24 and 72 hr. No such effect was detected in the human fibroblast cell line. ANIMAL STUDIES: Chronic administration to rats has caused tubular degeneration in the kidneys. Calcification and damage to the renal parenchyma occurred in a subchronic study in dogs. Developmental studies have been conducted in mice, rats and rabbits. An increase in the incidence and severity of hydronephrosis (distention of the renal pelvis and occasionally the ureters) was seen in a mouse study and one of three rabbit studies. In male mice treated intraperitoneally with furosemide at 0.3-50 mg/kg bw, a non-dose-dependent increase in the percentage of meiotic cells with chromosomal aberrations was observed during the whole spermatogenic cycle. Furosemide was tested over a wide range of doses (0, 100, 333, 1000, 3333, and 10,000 ug/plate) in four Salmonella typhimurium strains (TA98, TA100, TA1535, and TA1537) with and without metabolic activation. Furosemide was negative in these tests and the highest ineffective dose level tested in any Salmonella tester strain was 10,000 ug/plate. Furosemide was reported to induce mutations in L5178Y mouse lymphoma cells in the presence of an exogenous metabolic system only at the highest concentration tested (1500 ug/mL). It was also reported to induce sister chromatid exchange and chromosomal aberrations in Chinese hamster CHO cells at 3750 and 500 ug/mL in the presence and absence of an exogenous metabolic system. Furosemide induced chromosomal damage in Chinese hamster lung fibroblasts in vitro, but only in the absence of an exogenous metabolic system. ECOTOXICITY STUDIES: Furosemide in combination with other drugs found in waste water was genotoxic to zebrafish, and exhibited toxic effects on riverine microbial communities. Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Because little information is available on the use of furosemide during breastfeeding and because intense diuresis from high doses might decrease lactation, an alternate drug may be preferred, especially while nursing a newborn or preterm infant. Low doses of furosemide (20 mg daily) do not suppress lactation. ◉ Effects in Breastfed Infants Anecdotal, short-term observations at one medical center found no adverse infant effects from maternal use of furosemide in the immediate postpartum period. ◉ Effects on Lactation and Breastmilk Furosemide 20 mg intramuscularly on the first postpartum day followed by 40 mg orally for 4 days has been used in conjunction with fluid restriction and breast binding to suppress lactation within 3 days postpartum. The added contribution of furosemide to fluid restriction and breast binding, which are effective in suppressing lactation, is not known. No data exist on the effects of loop diuretics on established lactation. A randomized, controlled trial compared postpartum furosemide (n = 192) to placebo (n = 192) in women who had gestational hypertension and preeclampsia. Patients received either a 4- to 5-day course of 20 mg oral furosemide daily or placebo. The first dose was given 6 to 24 hours postpartum and then every 24 hours thereafter until hospital discharge. No difference was found in patient-reported breastfeeding difficulties between the two groups. A study of mothers with antepartum hypertension were given either furosemide 20 mg or a placebo daily for 5 days postpartum. Mothers reported whether they were exclusively breastfeeding at 2 and 6 weeks postpartum. No difference was found in the rates of exclusive breastfeeding between the furosemide and placebo groups. Protein Binding Plasma concentrations ranging from 1 to 400 mcg/mL are about 91-99% bound in healthy individuals. The unbound fraction is about 2.3-4.1% at therapeutic concentrations. Furosemide mainly binds to serum albumin. - Ototoxicity in mice: Intraperitoneal administration of Furosemide sodium (400 mg/kg) combined with kanamycin (400 mg/kg) induced irreversible hearing loss in mice, characterized by increased ABR thresholds and reduced outer hair cell count in the cochlea (histological analysis showed 40%–50% outer hair cell loss in the basal turn)[3] - Cytotoxicity in human cells: Furosemide sodium (up to 200 μM) had no significant cytotoxicity on normal human gastric epithelial cells or human erythrocytes (cell viability > 90% after 72-hour incubation), but exhibited selective cytotoxicity on poorly differentiated human gastric cancer cells (48% viability reduction at 200 μM)[4,5] |
| References |
[1]. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J Biol Chem. 1996 Jul 5;271(27):16237-44. [2]. Residues in transmembrane domains I and II determine gamma-aminobutyric acid type AA receptor subtype-selective antagonism by Furosemide sodium. Mol Pharmacol. 1999 Jun;55(6):993-9. [3]. Novel Peptide Vaccine GV1001 Rescues Hearing in Kanamycin/Furosemide sodium-Treated Mice. Front Cell Neurosci. 2018 Jan 19;12:3. [4]. Furosemide sodium, a blocker of Na+/K+/2Cl- cotransporter, diminishes proliferation of poorly differentiated human gastric cancer cells by affecting G0/G1 state. J Physiol Sci. 2006 Dec;56(6):401-6. [5]. Inhibitory effect of Furosemide sodium on non-selective voltage-independent cation channels in human erythrocytes.Cell Physiol Biochem. 2012;30(4):863-75. |
| Additional Infomation |
Furosemide is an odorless white to slightly yellow crystalline powder. A diuretic drug. Almost tasteless. (NTP, 1992) National Toxicology Program, Institute of Environmental Health Sciences, National Institutes of Health (NTP). 1992. National Toxicology Program Chemical Repository Database. Research Triangle Park, North Carolina. Furosemide is a chlorobenzoic acid that is 4-chlorobenzoic acid substituted by a (furan-2-ylmethyl)amino and a sulfamoyl group at position 2 and 5 respectively. It is a diuretic used in the treatment of congestive heart failure. It has a role as a xenobiotic, an environmental contaminant and a loop diuretic. It is a sulfonamide, a chlorobenzoic acid and a member of furans. Furosemide is a potent loop diuretic that acts on the kidneys to ultimately increase water loss from the body. It is an anthranilic acid derivative. Furosemide is used for edema secondary to various clinical conditions, such as congestive heart failure exacerbation, liver failure, renal failure, and high blood pressure. It mainly works by inhibiting electrolyte reabsorption from the kidneys and enhancing the excretion of water from the body. Furosemide has a fast onset and short duration of action and has been used safely and effectively in both pediatric and adult patients. The use of furosemide is particularly beneficial in clinical settings that require a drug with a higher diuretic potential. In addition to oral formulations, the solution for intravenous and intramuscular administration is also available, which is typically limited to patients who are unable to take oral medication or for patients in emergency clinical situations. Furosemide is a Loop Diuretic. The physiologic effect of furosemide is by means of Increased Diuresis at Loop of Henle. Furosemide is a sulfamoylanthranilic acid derivative, also known as frusemide, and potent loop diuretic. Furosemide is widely used to treat hypertension and edema. This agent is highly bound to albumin and is largely excreted unchanged in the urine. A benzoic-sulfonamide-furan. It is a diureti. Drug Indication Furosemide is indicated for the treatment of edema associated with congestive heart failure, cirrhosis of the liver, and renal disease, including the nephrotic syndrome, in adults and pediatric patients. Oral furosemide is indicated alone for the management of mild to moderate hypertension or severe hypertension in combination with other antihypertensive medications. Intravenous furosemide is indicated as adjunctive therapy in acute pulmonary edema when a rapid onset of diuresis is desired. Subcutaneous furosemide is indicated for the treatment of congestion due to fluid overload in adults with NYHA Class II/III chronic heart failure. This drug formulation is not indicated for emergency situations or in patients with acute pulmonary edema. View MoreTherapeutic UsesDiuretics; Sodium Potassium Chloride Symporter Inhibitors Oral Lasix may be used in adults for the treatment of hypertension alone or in combination with other antihypertensive agents. Hypertensive patients who cannot be adequately controlled with thiazides will probably also not be adequately controlled with Lasix alone. /Included in US product labeling/ Lasix is indicated in adults and pediatric patients for the treatment of edema associated with congestive heart failure, cirrhosis of the liver, and renal disease, including the nephrotic syndrome. Lasix is particularly useful when an agent with greater diuretic potential is desired. /Included in US product labeling/ IV furosemide has been found useful as an adjunct to hypotensive agents in the treatment of hypertensive crises, especially when associated with acute pulmonary edema or renal failure. In addition to producing a rapid diuresis, furosemide enhances the effects of other hypotensive drugs and counteracts the sodium retention caused by some of these agents. Drug Warnings /BOXED WARNING/ Lasix (furosemide) is a potent diuretic which, if given in excessive amounts, can lead to a profound diuresis with water and electrolyte depletion. Therefore, careful medical supervision is required and dose and dose schedule must be adjusted to the individual patient's needs. Excessive diuresis may cause dehydration and blood volume reduction with circulatory collapse and possibly vascular thrombosis and embolism, particularly in elderly patients. As with any effective diuretic, electrolyte depletion may occur during Lasix therapy, especially in patients receiving higher doses and a restricted salt intake. Hypokalemia may develop with Lasix, especially with brisk diuresis, inadequate oral electrolyte intake, when cirrhosis is present, or during concomitant use of corticosteroids, ACTH, licorice in large amounts, or prolonged use of laxatives. Digitalis therapy may exaggerate metabolic effects of hypokalemia, especially myocardial effects. Patients receiving furosemide must be carefully observed for signs of hypovolemia, hyponatremia, hypokalemia, hypocalcemia, hypochloremia, and hypomagnesemia. Patients should be informed of the signs and symptoms of electrolyte imbalance and instructed to report to their physicians if weakness, dizziness, fatigue, faintness, mental confusion, lassitude, muscle cramps, headache, paresthesia, thirst, anorexia, nausea, and/or vomiting occur. Excessive fluid and electrolyte loss may be minimized by initiating therapy with small doses, careful dosage adjustment, using an intermittent dosage schedule if possible, and monitoring the patient's weight. To prevent hyponatremia and hypochloremia, intake of sodium may be liberalized in most patients; however, patients with cirrhosis usually require at least moderate sodium restriction while on diuretic therapy. Determinations of serum electrolytes, BUN, and carbon dioxide should be performed early in therapy with furosemide and periodically thereafter. If excessive diuresis and/or electrolyte abnormalities occur, the drug should be withdrawn or dosage reduced until homeostasis is restored. Electrolyte abnormalities should be corrected by appropriate measures. Pharmacodynamics Furosemide manages hypertension and edema associated with congestive heart failure, cirrhosis, and renal disease, including the nephrotic syndrome. Furosemide is a potent loop diuretic that works to increase the excretion of Na+ and water by the kidneys by inhibiting their reabsorption from the proximal and distal tubules, as well as the loop of Henle. It works directly acts on the cells of the nephron and indirectly modifies the content of the renal filtrate. Ultimately, furosemide increases the urine output by the kidney. Protein-bound furosemide is delivered to its site of action in the kidneys and secreted via active secretion by nonspecific organic transporters expressed at the luminal site of action. Following oral administration, the onset of the diuretic effect is about 1 and 1.5 hours, and the peak effect is reached within the first 2 hours. The duration of effect following oral administration is about 4-6 hours but may last up to 8 hours. Following intravenous administration, the onset of effect is within 5 minutes, and the peak effect is reached within 30 minutes. The duration of action following intravenous administration is approximately 2 hours. Following intramuscular administration, the onset of action is somewhat delayed. Mechanism of Action Furosemide promotes diuresis by blocking tubular reabsorption of sodium and chloride in the proximal and distal tubules, as well as in the thick ascending loop of Henle. This diuretic effect is achieved through the competitive inhibition of sodium-potassium-chloride cotransporters (NKCC2) expressed along these tubules in the nephron, preventing the transport of sodium ions from the lumenal side into the basolateral side for reabsorption. This inhibition results in increased excretion of water along with sodium, chloride, magnesium, calcium, hydrogen, and potassium ions. As with other loop diuretics, furosemide decreases the excretion of uric acid. Furosemide exerts direct vasodilatory effects, which results in its therapeutic effectiveness in the treatment of acute pulmonary edema. Vasodilation leads to reduced responsiveness to vasoconstrictors, such as angiotensin II and noradrenaline, and decreased production of endogenous natriuretic hormones with vasoconstricting properties. It also leads to increased production of prostaglandins with vasodilating properties. Furosemide may also open potassium channels in resistance arteries. The main mechanism of action of furosemide is independent of its inhibitory effect on carbonic anhydrase and aldosterone. Though both in vivo and in vitro studies have demonstrated an anticonvulsant effect of the loop diuretic furosemide, the precise mechanism behind this effect is still debated. The current study investigates the effect of furosemide on Cs-induced epileptiform activity (Cs-FP) evoked in area CA1 of rat hippocampal slices in the presence of Cs(+) (5mM) and ionotropic glutamatergic and GABAergic receptor antagonists. As this model diverges in several respects from other epilepsy models it can offer new insight into the mechanism behind the anticonvulsive effect of furosemide. The present study shows that furosemide suppresses the Cs-FP in a dose-dependent manner with a near complete block at concentrations = 1.25 mM. Because furosemide targets several types of ion transporters we examined the effect of more selective antagonists. Bumetanide (20 uM), which selectively inhibits the Na-K-2Cl co-transporter (NKCC1), had no significant effect on the Cs-FP. VU0240551 (10 uM), a selective antagonist of the K-Cl co-transporter (KCC2), reduced the ictal-like phase by 51.73 +/- 8.5% without affecting the interictal-like phase of the Cs-FP. DIDS (50 uM), a nonselective antagonist of Cl(-)/HCO3(-)-exchangers, Na(+)-HCO3(-)-cotransporters, chloride channels and KCC2, suppressed the ictal-like phase by 60.8 +/- 8.1% without affecting the interictal-like phase. At 500 uM, DIDS completely suppressed the Cs-FP. Based on these results we propose that the anticonvulsant action of furosemide in the Cs(+)-model is exerted through blockade of the neuronal KCC2 and Na(+)-independent Cl(-)/HCO3(-)-exchanger (AE3) leading to stabilization of the activity-induced intracellular acidification in CA1 pyramidal neurons. PMID:26301821 Sodium chloride reabsorption in the thick ascending limb of the loop of Henle is mediated by the Na(+)-K(+)-2Cl(-) cotransporter (NKCC2). The loop diuretic furosemide is a potent inhibitor of NKCC2. However, less is known about the mechanism regulating the electrolyte transporter. Considering the well-established effects of nitric oxide on NKCC2 activity, cGMP is likely involved in this regulation. cGMP-dependent protein kinase I (cGKI; PKGI) is a cGMP target protein that phosphorylates different substrates after activation through cGMP. We investigated the potential correlation between the cGMP/cGKI pathway and NKCC2 regulation. We treated wild-type (wt) and cGKIa-rescue mice with furosemide. cGKIa-rescue mice expressed cGKIa only under the control of the smooth muscle-specific transgelin (SM22) promoter in a cGKI deficient background. Furosemide treatment increased the urine excretion of sodium and chloride in cGKIa-rescue mice compared to that in wt mice. We analyzed the phosphorylation of NKCC2 by western blotting and immunostaining using the phosphospecific antibody R5. The administration of furosemide significantly increased the phosphorylated NKCC2 signal in wt but not in cGKIa-rescue mice. NKCC2 activation led to its phosphorylation and membrane translocation. To examine whether cGKI was involved in this process, we analyzed vasodilator-stimulated phosphoprotein, which is phosphorylated by cGKI. Furosemide injection resulted in increased vasodilator-stimulated phosphoprotein phosphorylation in wt mice. We hypothesize that furosemide administration activated cGKI, leading to NKCC2 phosphorylation and membrane translocation. This cGKI-mediated pathway could be a mechanism to compensate for the inhibitory effect of furosemide on NKCC2. - Furosemide sodium is a loop diuretic that exerts its physiological effect by inhibiting ion cotransporters (e.g., NKCC, KCC) in renal tubules[1,4] - The subtype-selective antagonism of Furosemide sodium on GABAAA receptors is mediated by residues in transmembrane domains I and II of the receptor’s α6 subunit[2] - Furosemide sodium-induced ototoxicity is often synergistic with aminoglycoside antibiotics (e.g., kanamycin), as shown by enhanced hearing loss in the combined treatment group compared to single-drug treatment[3] - The inhibition of NS-VICC by Furosemide sodium in human erythrocytes suggests a potential role in regulating erythrocyte volume and cation homeostasis[5] |
Solubility Data
| Solubility (In Vitro) |
DMSO : ≥ 150 mg/mL (~425.25 mM) H2O : ~100 mg/mL (~283.50 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (7.09 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (7.09 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (7.09 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 100 mg/mL (283.50 mM) in PBS (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.8350 mL | 14.1751 mL | 28.3503 mL | |
| 5 mM | 0.5670 mL | 2.8350 mL | 5.6701 mL | |
| 10 mM | 0.2835 mL | 1.4175 mL | 2.8350 mL |