Physicochemical Properties
| Molecular Formula | C12H15NO5S |
| Molecular Weight | 285.3162 |
| Exact Mass | 285.067 |
| CAS # | 122547-49-3 |
| Related CAS # | Faropenem;106560-14-9 |
| PubChem CID | 65894 |
| Appearance | Light yellow to yellow solid powder |
| Boiling Point | 570.2ºC at 760 mmHg |
| Flash Point | 298.7ºC |
| Vapour Pressure | 2.23E-15mmHg at 25°C |
| LogP | 0.3 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 3 |
| Heavy Atom Count | 19 |
| Complexity | 477 |
| Defined Atom Stereocenter Count | 4 |
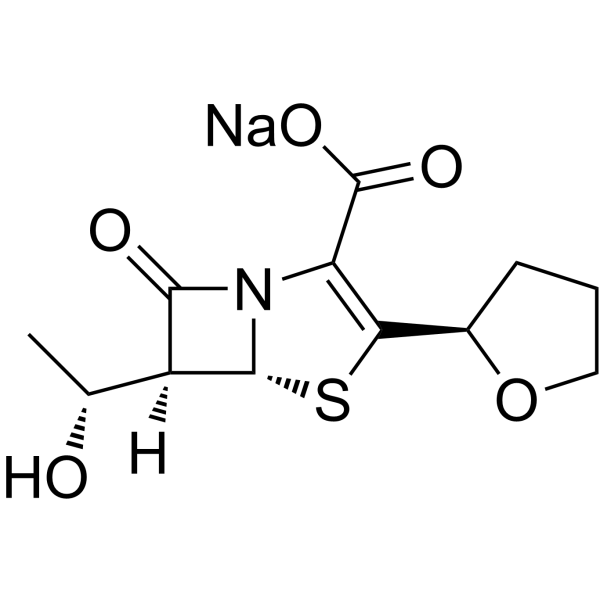
| SMILES | C[C@H]([C@@H]1[C@@H]2N(C1=O)C(=C(S2)[C@H]3CCCO3)C(=O)O)O |
| InChi Key | HGGAKXAHAYOLDJ-FHZUQPTBSA-N |
| InChi Code | InChI=1S/C12H15NO5S/c1-5(14)7-10(15)13-8(12(16)17)9(19-11(7)13)6-3-2-4-18-6/h5-7,11,14H,2-4H2,1H3,(H,16,17)/t5-,6-,7+,11-/m1/s1 |
| Chemical Name | (5R,6S)-6-[(1R)-1-hydroxyethyl]-7-oxo-3-[(2R)-oxolan-2-yl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
Faropenem targets the L,D-transpeptidases (LdtMt1 to LdtMt5) in Mycobacterium tuberculosis, which are enzymes responsible for the cross-linking of peptidoglycan. Kinetic constants (kcat/Kapp) for inactivation are provided: LdtMt1: 590 ± 170 µM⁻¹ min⁻¹; LdtMt2: 11 ± 0.3 µM⁻¹ min⁻¹; LdtMt3: 61 ± 3.9 µM⁻¹ min⁻¹; LdtMt4: 44 ± 3.3 µM⁻¹ min⁻¹; LdtMt5: No acylation detected. Faropenem also potentially targets D,D-carboxypeptidases and classical D,D-transpeptidases in M. tuberculosis. [1] |
| ln Vitro |
The Minimum Inhibitory Concentration (MIC) of faropenem against Mycobacterium tuberculosis H37Rv was determined to be 1.3 µg/ml, both in the presence and absence of the β-lactamase inhibitor clavulanate. [1] In time-kill kinetic assays, a single dose of faropenem (8 µg/ml, ~6x MIC) resulted in sustained killing of M. tuberculosis over 8 days. Increasing the dose to 28 µg/ml (~21x MIC) further enhanced killing. The bactericidal activity of faropenem was independent of clavulanate supplementation. [1] Faropenem (100 µg/ml) was found to be stable in 7H9 medium over 48 hours at 37°C, with little degradation observed by HPLC-UV analysis. [1] |
| ln Vivo |
In a murine acute tuberculosis model (C57BL/6J mice infected intratracheally with M. tuberculosis H37Rv), treatment with faropenem medoxomil (500 mg/kg) alone did not significantly reduce bacterial loads in the lungs. However, the combination of faropenem medoxomil (500 mg/kg), clavulanate (25 mg/kg), and probenecid (200 mg/kg), administered orally four times daily for 7 days, resulted in a modest but statistically significant reduction in pulmonary bacterial load compared to untreated controls (P < 0.05, Mann-Whitney test). [1] |
| Enzyme Assay |
The kinetic constants for the inactivation of the M. tuberculosis L,D-transpeptidase LdtMt1 by faropenem were determined. The reaction was carried out in 100 mM sodium phosphate buffer (pH 6.0) at 10°C. Inactivation was monitored by measuring the decrease in absorbance at 299 nm resulting from the opening of the β-lactam ring. The first-order rate constant (kobs) was determined for six antibiotic concentrations. The maximal acylation rate constant (kinact) and the apparent dissociation constant (Kapp) were deduced by fitting the data to the equation kobs = kinact[I]/(Kapp+[I]), where [I] is the antibiotic concentration. The hydrolysis rate constant (khydro) of the acyl enzyme was determined by incubating faropenem (100 µM) with increasing concentrations of LdtMt1 (0, 2.5, 5, and 10 µM) in the same buffer at 20°C and measuring the absorbance decrease at 299 nm over a longer time scale (500 min). Enzyme turnover (khydro) was calculated from the slope of the plot of hydrolysis velocity versus enzyme concentration. [1] The hydrolysis of faropenem by the M. tuberculosis β-lactamase BlaC was also assessed. The velocity of hydrolysis was determined at 258 nm in 100 mM MES buffer (pH 6.4) at 20°C over 500 minutes using various enzyme concentrations (0.17, 0.35, or 1 µM). Kinetic parameters (kcat and Km) were determined by fitting the velocity data against substrate concentration to the Michaelis-Menten equation. [1] |
| Cell Assay |
The MIC of faropenem was determined using a luciferase-based assay. M. tuberculosis Erdman expressing bacterial luciferase was grown to mid-log phase, diluted, and added to 96-well plates containing serial dilutions of faropenem (0.0015 to 200 µg/ml) with or without 5 µg/ml clavulanate. Bioluminescence was measured after 4 and 7 days of incubation at 37°C. The MIC was defined as the lowest concentration causing a 90% reduction in bioluminescence compared to the no-drug control. [1] For intracellular killing assays, RAW macrophages were infected with GFP-expressing M. tuberculosis. After infection and washing, the cells were treated with faropenem (7, 28, or 56 µg/ml). The medium containing the antibiotic was changed every 24 hours. At various time points, macrophages were lysed, and the lysates were serially diluted and plated on agar to enumerate bacterial CFU after 3-4 weeks of incubation. The fraction of infected macrophages was also analyzed by flow cytometry. [1] Real-time single-cell analysis was performed using time-lapse microscopy and microfluidics. An M. tuberculosis strain expressing GFP was cultured in a microfluidic device under a constant flow of medium. After a pre-antibiotic period, the flow medium was switched to one containing faropenem (28 µg/ml). Phase-contrast and fluorescence images were captured at regular intervals (e.g., 1-hour). Single-cell growth rates, division, and cytolysis (scored as abrupt loss of GFP fluorescence and phase intensity) were tracked and quantified using image analysis software. [1] |
| Animal Protocol |
Adult female C57BL/6J mice were infected with M. tuberculosis H37Rv (1 x 10⁵ CFU) via the intratracheal route. Treatment commenced on day 1 post-infection. Mice were treated with faropenem medoxomil (500 mg/kg), clavulanate (25 mg/kg), and probenecid (200 mg/kg), administered orally (by gavage) four times a day for 7 consecutive days, with a final dose on day 8. The compounds were formulated in 1% methylcellulose in PBS. On day 9 post-infection, the lungs were harvested, homogenized, and plated on 7H10 agar to enumerate bacterial CFU after 2-3 weeks of incubation. [1] |
| ADME/Pharmacokinetics |
In mice, after a single oral administration of faropenem (500 mg/kg) formulated in 1% methylcellulose/PBS, the pharmacokinetic parameters were: Tmax 0.25 hours, Cmax 69.6 ± 16.1 µg/ml, AUC 65.9 ± 19.1 µg·h/ml. When co-administered with probenecid (200 mg/kg), the parameters were: Tmax 0.25 hours, Cmax 83.8 ± 13.4 µg/ml, AUC 126.2 ± 20.5 µg·h/ml. When co-administered with probenecid and clavulanate (25 mg/kg), the parameters were: Tmax 0.25 hours, Cmax 83.1 ± 15.6 µg/ml, AUC 107.1 ± 38.7 µg·h/ml. [1] Faropenem is described as an orally bioavailable penem antibiotic (72-84% bioavailability in humans, based on the introduction). [1] |
| References |
[1]. Rapid cytolysis of Mycobacterium tuberculosis by faropenem, an orally bioavailable β-lactam antibiotic. Antimicrob Agents Chemother. 2015 Feb;59(2):1308-19. |
| Additional Infomation |
6alpha-[(R)-1-hydroxyethyl]-2-[(R)-tetrahydrofuran-2-yl]pen-2-em-3-carboxylic acid is a faropenem. It is a conjugate acid of a 6alpha-[(R)-1-hydroxyethyl]-2-[(R)-tetrahydrofuran-2-yl]pen-2-em-3-carboxylate. Faropenem has been used in trials studying the treatment of Tuberculosis, Pulmonary Tuberculosis, and Community Acquired Pneumonia. Faropenem is a penem with a tetrahydrofuran substituent at position C2, with broad-spectrum antibacterial activity against many gram-positive and gram-negative aerobes and anaerobes. Compared with imipenem, faropenem has improved chemical stability and reduced central nervous system effects. In addition, faropenem is resistant to hydrolysis by many beta-lactamases. Faropenem is an orally bioavailable penem antibiotic, more resistant to hydrolysis by β-lactamases than cephalosporins and carbapenems. It has been approved for human use for other indications. [1] The study identifies faropenem as a promising candidate for repurposing against drug-resistant tuberculosis (MDR-/XDR-TB). [1] Using real-time single-cell analysis, faropenem was shown to induce rapid and sustained growth arrest in M. tuberculosis followed by cytolysis. The rate of cytolysis by faropenem (kfast ~0.049 h⁻¹) was about 2-fold higher than meropenem-clavulanate and about 20-fold higher than isoniazid during the initial killing phase. It was also superior in eliminating a subpopulation of non-growing but metabolically active (NGMA) cells. [1] Unlike its effect on E. coli, where cells continue to grow until lysis, faropenem causes immediate growth arrest in M. tuberculosis, suggesting a different mechanism of action in this pathogen. [1] |
Solubility Data
| Solubility (In Vitro) |
H2O : ~100 mg/mL (~325.41 mM) DMSO : ~25 mg/mL (~81.35 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (8.14 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (8.14 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (8.14 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: 100 mg/mL (325.41 mM) in PBS (add these co-solvents sequentially from left to right, and one by one), clear solution; with ultrasonication. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.5048 mL | 17.5242 mL | 35.0484 mL | |
| 5 mM | 0.7010 mL | 3.5048 mL | 7.0097 mL | |
| 10 mM | 0.3505 mL | 1.7524 mL | 3.5048 mL |