FX1 is a potent and specific inhibitor of the BCL6 (B cell lymphoma 6) with an IC50 of approximately 35 μM. FX1 is ten times more powerful than the endogenous corepressors and binds to a crucial portion of the BCL6 lateral groove. FX1 mimicked the phenotype of mice engineered to express BCL6 with corepressor binding site mutations and disrupted the formation of the BCL6 repression complex, reactivating BCL6 target genes. In mice with DLBCL xenografts, low doses of FX1 caused the regression of tumors that had already developed. In addition, ex vivo primary human ABC-DLBCL specimens as well as ABC-DLBCL cells grown in vitro and in vivo were suppressed by FX1. All three of the BCL6 target genes are markedly reduced in BCOR and SMRT recruitment by FX1, but not at a negative control locus.
Physicochemical Properties
| Molecular Formula | C14H9CLN2O4S2 | |
| Molecular Weight | 368.8153 | |
| Exact Mass | 367.969 | |
| Elemental Analysis | C, 45.59; H, 2.46; Cl, 9.61; N, 7.60; O, 17.35; S, 17.39 | |
| CAS # | 1426138-42-2 | |
| Related CAS # |
|
|
| PubChem CID | 135886621 | |
| Appearance | Red solid powder | |
| LogP | 1.4 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 6 | |
| Rotatable Bond Count | 4 | |
| Heavy Atom Count | 23 | |
| Complexity | 808 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | 0 |
|
| InChi Key | JYBGCTWNOMSQJY-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C14H9ClN2O4S2/c15-6-1-2-8-7(5-6)10(12(20)16-8)11-13(21)17(14(22)23-11)4-3-9(18)19/h1-2,5,21H,3-4H2,(H,18,19) | |
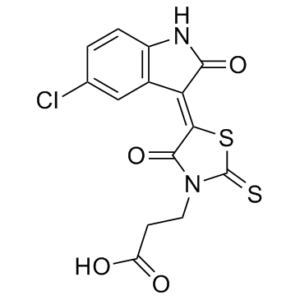
| Chemical Name | 3-[5-(5-chloro-2-oxoindol-3-yl)-4-hydroxy-2-sulfanylidene-1,3-thiazol-3-yl]propanoic acid | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | BCL6 (IC50 ~35 μM) |
| ln Vitro | For 30 minutes, DLBCL cells are exposed to 50 μM FX1. All three of the BCL6 target genes are markedly reduced in BCOR and SMRT recruitment by FX1, but not at a negative control locus. The BCL6-negative DLBCL cell line, which is unaffected by FX1, has little SMRT at these loci. When FX1 and 79-6 are pitted against one another in quantitative ChIP assays in DLBCL cells after treatment with 50 μM FX1 for 6 hours, it is clear that FX1 has greater potency than 79-6 in preventing BCL6 binding to SMRT. mRNA is collected at four sequential time points after FX1 exposure to DLBCL cells. In two separate DLBCL cell lines, FX1 almost always significantly induces derepression of these genes when compared with vehicle[1]. |
| ln Vivo |
Spleens in FX1-treating mice are macroscopically indistinguishable from vehicle controls. TFX1 has no impact on the overall abundance of B cells as determined by flow cytometry. When exposed to FX1, GC B cells (GL7+FAS+B220+) are significantly reduced. IHC analyzes the architecture of the spleen. Normal B cell follicular structures can be seen after staining with the B220 antibody, but a significant loss of GCs can be seen after staining with the GC-specific B cell marker peanut agglutinin. The half-life is thought to be 12 hours or so. Finally, it is determined if FX1 can cause toxic effects in mice. H&E-stained sections of the lung, digestive tract, heart, kidney, liver, spleen, and bone marrow of mice treated with FX1 compared with vehicle show no signs of toxicity, inflammation, or infection[1]. FX1 phenocopies the BCL6 BTB domain phenotype in vivo. [1] Mutation of the BCL6 BTB corepressor binding site results in normal B cell development but profound loss of GC formation in mice, with only small residual GCs forming after T cell–dependent immunization (e.g., as shown in Supplemental Figure 3A and ref. 22). To determine whether FX1 could recapitulate this phenotype, Researchers immunized C57BL/6 mice with sheep red blood cells, a T cell–dependent antigen, and then treated them with daily doses of FX1 at 80 mg/kg or vehicle. After 10 days of treatment (when GCs are normally at their peak), mice were euthanized and spleens were collected. Spleens in FX1-treated mice were macroscopically indistinguishable from vehicle controls (Figure 3A). As expected, total B cell abundance measured by flow cytometry was unaffected by FX1 (Figure 3B). In contrast and similar to the BCL6 BTB mutant phenotype, GC B cells (GL7+FAS+B220+) were significantly depleted by exposure to FX1 (P < 0.0001; Figure 3C). Researchers also examined splenic architecture by IHC. Staining with B220 antibody revealed normal B cell follicular structures, whereas staining for the GC B cell–specific marker peanut agglutinin showed profound loss of GCs (Figure 3D). This defect was further manifest by the significant reduction in the number of GCs (P = 0.0001) and the spleen surface area occupied by GCs (2.4-fold; P < 0.001) in comparison with vehicle control (Figure 3D). Loss of proliferating GC B cells was also evident through Ki-67 staining, which showed loss of Ki-67+ GC structures in FX1- versus vehicle-treated mice (Figure 3D). FX1 potently suppresses BCL6-dependent GCB-DLBCLs in vitro and in vivo. [1] The purpose of FX1 is to kill BCL6-dependent tumors. BCL6 is highly expressed in the GCB class of DLBCLs. DLBCL cell lines have been previously classified as BCL6 dependent or independent on the basis of whether they are affected by BCL6 knockdown or inhibition (34). To assess the capacity of FX1 to suppress DLBCLs, Researchers treated a panel of GCB-DLBCL cell lines (8 BCL6 dependent and 4 BCL6 independent) with different concentrations of FX1 for 48 hours and determined the concentration of compound required to inhibit 50% of growth in comparison with vehicle-treated cells (GI50). FX1 showed a selective effect on BCL6-dependent DLBCLs with average GI50 values of about 36 μM, whereas GI50 values could not be determined in BCL6-independent DLBCLs, since they did not even reach 50% growth inhibition at concentrations of drug higher than 125 μM (Figure 4A). It should be noted that some of the DLBCL cell lines defined as BCL6 independent still retain a small degree of responsiveness to BCL6 inhibitors (34). The mechanism through which loss of BCL6 dependency occurs is not known, although in certain cases (e.g., Toledo cells) this is accompanied by almost complete loss of BCL6 expression (Supplemental Figure 4A). [1] Researchers next wished to assess the antilymphoma activity of FX1 in vivo. However, Researchers first assessed the basic pharmacokinetics of the compound. Sixteen SCID mice bearing SUDHL-6 xenografts were given 50 mg/kg of FX1 via i.p. injection, followed by serial collection of serum and tissue specimens. FX1 was observed to maintain a serum concentration of 50 μM for 10 hours (Supplemental Figure 4B). The half-life was estimated to be approximately 12 hours (Supplemental Figure 4B). mRNA extracted from tumor tissue from these mice was assessed for abundance of BCL6 target genes CD69, CDKN1A, and CXCR4. Maximal induction of target genes occurred at 4–6 hours and subsequently declined (Supplemental Figure 4C). Finally, Researchers assessed whether FX1 could induce toxic effects in mice. BCL6 BTB mutant mice have no organ dysfunction or illness, and so Researchers would not expect on-target toxicity (22). A cohort of 10 BALB/c mice were treated for 10 days with daily doses of 100 mg/kg/d FX1, after which the animals were sacrificed and analyzed for histological evidence of organ damage (Supplemental Figure 4D and Supplemental Table 2). No signs of toxicity, inflammation, or infection were evident from H&E-stained sections of lung, gastrointestinal tract, heart, kidney, liver, spleen, and bone marrow of the fixed organs from mice treated with FX1 compared with vehicle. Researchers also examined peripheral blood counts and serum chemistry in FX1-treated mice, all of which remained within normal parameters (Supplemental Table 2). [1] To determine efficacy of FX1 as compared with 79-6 in vivo, Researchers first established DLBCL xenografts using OCI-Ly7 DLBCL cells in SCID mice. When tumors reached a volume of approximately 100 mm3, treatment was initiated with 25 mg/kg or 50 mg/kg of FX1 or 79-6 daily, or vehicle. Animals were sacrificed when vehicle controls reached maximal permitted tumor burden (at day 10). Strikingly, FX1 caused profound and significant suppression of DLBCLs (P = 0.001; Figure 4B), and indeed not only prevented growth of the xenografts but in addition caused these tumors to shrink from their initial volume. The maximal effect was already achieved by the lower 25 mg/kg dose. In contrast, 79-6 exhibited much weaker antitumor activity at these doses (42% reduction in tumor growth at 25 mg/kg and 64% with 50 mg/kg) (Figure 4C). TUNEL and Ki-67 staining showed that FX1 also induced more apoptosis and growth arrest than 79-6, respectively (Supplemental Figure 4E). This potent dose-dependent antilymphoma effect of FX1 was confirmed in a second xenograft model using SUDHL-6 DLBCL cells (Supplemental Figure 4, F and G). To confirm that antitumor effects were lymphoma cell autonomous and not due to effects on the host, Researchers also tested the effect of FX1 on BCL6-independent Toledo xenografts. Exposure to 50 mg/kg FX1 had no effect on these xenografts (Supplemental Figure 4, H–J). [1] To verify that ABC-DLBCLs can also be targeted by FX1 in vivo, Researchers generated xenografts using the HBL-1 ABC-DLBCL cell line suspended in Matrigel s.c. in NOD/SCID mice. When tumors reached about 100 mm3, Researchers treated the mice with daily doses of 50 mg/kg FX1. After 10 days of treatment Researchers observed an approximately 70% decrease in the tumor volume of mice exposed to 50 mg/kg FX1 (Figure 5, B and C). Researchers also observed an increase in the apoptotic cells from 2.5% to 10% by TUNEL staining in FX1-treated tumors compared with those treated with vehicle (Figure 5D). [1] |
| Enzyme Assay |
MST measurements. [1] Recombinant BCL6 BTB was labeled using the RED-NHS Labeling kit. The labeling reaction was performed according to the manufacturer’s instructions in the supplied labeling buffer applying a concentration of 20 μM protein (molar dye/protein ratio ~2:1) at room temperature for 30 minutes. Unreacted dye was removed with the supplied dye removal columns equilibrated with PBS buffer (PBS, 0.005% Tween-80). The label/protein ratio was determined using photometry at 650 nm and Bradford reagent. Thereby, a ratio of 0.8 was typically achieved. The labeled BCL6 BTB was adjusted to 400 nM with PBS buffer supplemented with 0.05% Tween-80. SMRT, 79-6, and FX1 were dissolved in PBS buffer supplemented with 0.05% Tween-80 and 10% DMSO, and a series of 16 1:1 dilutions were prepared in the identical buffer, producing ligand concentrations ranging from 19 pM to 625 μM. For thermophoresis, each ligand dilution was mixed with 1 volume of labeled BCL6 BTB, which leads to a final concentration of fluorescently labeled BCL6 BTB of 200 nM and final ligand concentrations ranging from 9 pM to 312 nM in a 5% DMSO final concentration. After 10 minutes of incubation, approximately 4 μl of each solution was filled into Monolith NT Standard Treated Capillaries. Thermophoresis was measured using a Monolith NT.115 instrument at a temperature of 25°C with 5 seconds/30 seconds/5 seconds laser off/on/off times, respectively. Instrument parameters were adjusted with 90% light-emitting diode (LED) power and 40% MST power. Data of 3 independent experiments were analyzed using the signal from thermophoresis. NMR experiments. [1] All NMR experiments were recorded at 30°C using a Bruker 600 MHz spectrometer equipped with cryogenic probe. Assignment of BCL6 BTB domain was performed based on HNCA and HNCOCA experiments recorded for 200 μM 13C, 15N BCL6, and previously published assignment. Fo-Fc electron density maps found in the lateral groove site of the BCL6 BTB contoured at 2.0 σ level. Due to insufficient occupancy, full structure refinement of the 1085 in the complex could not be reliably completed. |
| Cell Assay |
Using antibodies for BCL6, SMRT, BCOR, or IgG control, quantitative ChIP is carried out in SUDHL-6 cells exposed to FX1 (black bars) or vehicle (white bars) in DLBCL cells in order to enrich for known BCL6 binding sites in the CD69, CXCR4, and DUSP5 loci, as well as a negative control region.
Combination of FX1 with doxorubicin. [1] For combination treatments, we exposed cells to a dose curve of each drug alone or their combination in constant ratio, and cell viability was determined by CellTiter-Blue. To compare different schedules of treatments, we treated the cells in triplicate as follows: FX1 and doxorubicin simultaneously and cells treated for 48 hours; FX1 first and 24 hours after we added doxorubicin and treated for an extra 48 hours; doxorubicin first and 24 hours after we added FX1 and treated for an extra 48 hours. Then, we used CompuSyn software to plot dose-effect curves and calculate the dose-reduction index. Primary cell treatment. [1] Single-cell suspensions from lymph node biopsies were thawed and resuspended in Advanced RPMI supplemented with 20% human serum, GlutaMAX (2X), glycine (5 mM), and penicillin and streptomycin. Cell number and viability were determined by counting with trypan blue. Irradiated HK cells (2,000 rad) cultured in DMEM supplemented with 10% FBS and penicillin and streptomycin adhered to tissue culture plates at 37°C. Medium was aspirated and patient samples were plated on the HK feeder layer. Samples were exposed to FX1 or vehicle for 48 hours and analyzed for viability by staining with PerCP Cy5.5-conjugated anti-CD3 and FITC-conjugated anti-CD20 as well as annexin V and DAPI. CD20+CD3– cells that were annexin V/DAPI double negative were considered live, malignant B cells. Primary samples were classified by Hans IHC criteria. |
| Animal Protocol |
SCID mice bearing SUDHL-6 xenografts 50 mg/kg i.p. Six- to 8-week-old male SCID mice were injected s.c. with 10~7 low-passage human SUDHL-6, OCI-Ly7, or Toledo cells. Alternatively, 6- to 8-week-old NOD/SCID mice were injected with low-passage HBL-1 cells. When tumors reached a palpable size (approximately 100 mm3), mice were assigned in a randomized way to treatment groups and treated i.p. with 25 or 50 mg/kg/d of the drugs. Drugs were reconstituted in PEG-400 and stored at –20°C until use. Tumor size was measured 3 times a week with an electronic digital caliper in 2 dimensions, and then tumor volume was calculated with the formula: tumor volume (mm3) = (smallest diameter2 × largest diameter)/2. Data are expressed as mean ± SEM, and differences were considered statistically significant at P less than 0.05 by Mann-Whitney test. [1] |
| References |
[1]. Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. J Clin Invest. 2016 Sep 1; 126(9): 3351–3362. |
| Additional Infomation |
FX1 is a member of the class of oxindoles that is 5-chloro-oxindole in which the methylene hydrogens at position 3 have been replaced by an N-(2-carboxyethyl)rhodanin-5-ylidene group. It has a role as an antineoplastic agent. It is a member of oxindoles, an organochlorine compound, a thiazolidinone and a monocarboxylic acid. It is functionally related to a 3-methyleneoxindole and a rhodanine. Diffuse large B cell lymphomas (DLBCLs) arise from proliferating B cells transiting different stages of the germinal center reaction. In activated B cell DLBCLs (ABC-DLBCLs), a class of DLBCLs that respond poorly to current therapies, chromosomal translocations and amplification lead to constitutive expression of the B cell lymphoma 6 (BCL6) oncogene. The role of BCL6 in maintaining these lymphomas has not been investigated. Here, we designed small-molecule inhibitors that display higher affinity for BCL6 than its endogenous corepressor ligands to evaluate their therapeutic efficacy for targeting ABC-DLBCL. We used an in silico drug design functional-group mapping approach called SILCS to create a specific BCL6 inhibitor called FX1 that has 10-fold greater potency than endogenous corepressors and binds an essential region of the BCL6 lateral groove. FX1 disrupted formation of the BCL6 repression complex, reactivated BCL6 target genes, and mimicked the phenotype of mice engineered to express BCL6 with corepressor binding site mutations. Low doses of FX1 induced regression of established tumors in mice bearing DLBCL xenografts. Furthermore, FX1 suppressed ABC-DLBCL cells in vitro and in vivo, as well as primary human ABC-DLBCL specimens ex vivo. These findings indicate that ABC-DLBCL is a BCL6-dependent disease that can be targeted by rationally designed inhibitors that exceed the binding affinity of natural BCL6 ligands. [1] In many cell lines it appears that response to BCL6 inhibitors correlates with BCL6 levels, in that higher doses are required to suppress cell lines that express more BCL6. For example, OCI-Ly1 cells express high levels of BCL6 and need more compound than SUDHL-6 and DOHH-2 cells, which express less BCL6. However, there are many exceptions to this rule, likely due to the effects of different sets of somatic mutations and other factors that may alter BCL6 functionality. The study of primary DLBCLs is limited by the fact that these tumors cannot be maintained in culture long enough to fully assess the therapeutic impact of FX1. Yet our studies in primary specimens serve as proof of principle confirming cell line data showing that GCB- and ABC-DLBCL cells are BCL6 dependent and amenable to BCL6 inhibitors. Finally, FX1 could enhance the response of both GCB- and ABC-DLBCLs to cytotoxic therapy. It is therefore possible that combination of BCL6-targeted therapy with standard antilymphoma drugs could yield superior clinical efficacy, which would be of great interest especially in higher-risk patients who are more likely to be chemotherapy resistant. Hence our study expands the scope of lymphomas likely to respond to BCL6-targeted therapy to include both GCB- and ABC-type DLBCLs. This is particularly important given the more unfavorable outcome of ABC-DLBCLs. Efforts to further refine FX1 and generate additional chemical scaffolds using SILCS will be expected to yield compounds that could benefit these difficult-to-treat patients. [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 1 mg/mL (2.71 mM) (saturation unknown) in 10% DMSO + 40% PEG300 +5% Tween-80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 10.0 mg/mL clear DMSO stock solution to 400 μL of PEG300 and mix evenly; then add 50 μL of Tween-80 + to the above solution and mix evenly; then add 450 μL of normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.7113 mL | 13.5567 mL | 27.1135 mL | |
| 5 mM | 0.5423 mL | 2.7113 mL | 5.4227 mL | |
| 10 mM | 0.2711 mL | 1.3557 mL | 2.7113 mL |