FPS-ZM1 is a potent and high-affinity RAGE specific blocker with Ki value of 25 nM. It inhibits amyloid-β binding to RAGE, neurological damage and inflammation in the APP(sw/0) transgenic mouse model of AD. An increased level of advanced glycation end products (AGEs) is observed in brains of patients with Alzheimer's disease (AD). AGEs and receptor for AGEs (RAGE) play important roles in the pathogenesis of AD. FPS-ZM1 is not toxic to mice and can easily cross the blood-brain barrier. AGEs administration induced an-regulation of Abeta production, inflammation, and oxidative stress, and an increased escape latency of rats in the Morris water maze test, all of these are significantly reduced by FPS-ZM1 treatment. FPS-ZM1 might be a novel therapeutic agent to treat AD patients.
Physicochemical Properties
| Molecular Formula | C20H22CLNO | |
| Molecular Weight | 327.85 | |
| Exact Mass | 327.138 | |
| Elemental Analysis | C, 73.27; H, 6.76; Cl, 10.81; N, 4.27; O, 4.88 | |
| CAS # | 945714-67-0 | |
| Related CAS # |
|
|
| PubChem CID | 24752728 | |
| Appearance | White to off-white solid powder | |
| Density | 1.2±0.1 g/cm3 | |
| Boiling Point | 497.6±38.0 °C at 760 mmHg | |
| Flash Point | 254.7±26.8 °C | |
| Vapour Pressure | 0.0±1.3 mmHg at 25°C | |
| Index of Refraction | 1.603 | |
| LogP | 5.03 | |
| Hydrogen Bond Donor Count | 0 | |
| Hydrogen Bond Acceptor Count | 1 | |
| Rotatable Bond Count | 4 | |
| Heavy Atom Count | 23 | |
| Complexity | 366 | |
| Defined Atom Stereocenter Count | 0 | |
| InChi Key | XDFKWGIBQMHSOH-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C20H22ClNO/c21-18-13-11-17(12-14-18)20(23)22(19-9-5-2-6-10-19)15-16-7-3-1-4-8-16/h1,3-4,7-8,11-14,19H,2,5-6,9-10,15H2 | |
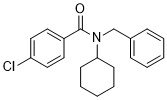
| Chemical Name | N-benzyl-4-chloro-N-cyclohexylbenzamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
RAGE (Ki = 25 nM)[1]
FPS-ZM1 targets the receptor for advanced glycation end products (RAGE) with a Ki value of 14 nM (human recombinant RAGE, SPR assay) [1] FPS-ZM1 specifically binds RAGE and inhibits its interaction with ligands (amyloid β [Aβ], advanced glycation end products [AGEs], HMGB1), with no significant binding to other receptors (e.g., TLR4, TNF-α receptor) [1][2][3] |
|
| ln Vitro |
Compared to its parent molecule FPS2, FPS-ZM1 inhibits Aβ/RAGE binding in CHO cells with an affinity that is about two times higher. Other recognized RAGE ligands, such as amphoterin and S100 calbindin B, are inhibited from binding to sRAGE by FPS-ZM1. When it comes to decreasing Aβ40-induced increases in BACE1 mRNA and protein levels as well as the generation of sAPPβ, the APP cleavage product of BACE1 that indicates BACE1 activity, FPS-ZM1 is more effective than FPS2 [1]. In human recombinant RAGE binding assays, FPS-ZM1 (0.1-100 nM) dose-dependently inhibited Aβ1-42 binding to RAGE, with an IC50 of 23 nM (fluorescence polarization assay, p < 0.01) [1] - In primary rat microglia stimulated with Aβ1-42 (10 μM), FPS-ZM1 (1-10 μM) reduced the release of pro-inflammatory cytokines TNF-α (48% reduction at 10 μM) and IL-6 (52% reduction at 10 μM) (ELISA, p < 0.05) [2] - FPS-ZM1 (1-10 μM) inhibited AGEs-induced reactive oxygen species (ROS) production in rat hippocampal neurons by 45% at 10 μM (DCFH-DA assay, p < 0.05) and reduced lipid peroxidation (malondialdehyde level decreased by 38%, p < 0.05) [2] - In SH-SY5Y neuroblastoma cells treated with Aβ1-42 (10 μM), FPS-ZM1 (3-30 μM) improved cell viability by 32% at 30 μM (MTT assay, p < 0.01) and reduced caspase-3 activation (western blot, 41% reduction at 30 μM) [1] - FPS-ZM1 (10 μM) blocked HMGB1-induced NF-κB activation in mouse microglia, as evidenced by reduced p65 nuclear translocation (immunofluorescence, p < 0.05) [3] |
|
| ln Vivo |
Mice cannot be poisoned by FPS-ZM1, and it easily penetrates the blood-brain barrier. In aged APPsw/0 mice overexpressing human Aβ precursor protein, an AD transgenic mouse model with established Aβ pathology, FPS-ZM1 inhibits RAGE-mediated influx of circulating Aβ40 into the brain and Aβ42 entry into the brain. FPS-ZM1 binds specifically to RAGE in the brain, suppressing microglial activation and neuroinflammatory responses while inhibiting β-secretase activity and Aβ production [1]. In AGEs rats, the levels of Aβ1-42 and Aβ1-40 were decreased by FPS-ZM1 treatment. It downregulates the AGEs-mediated increase in hippocampal pro-inflammatory cytokines and inhibits the AGEs-mediated increase in Aβ metabolism-related proteins. In rats with advanced ageing, FPS-ZM1 improves the antioxidant defense system and reduces the memory impairment caused by AGEs [2]. In APP/PS1 transgenic mice (Alzheimer disease model), intraperitoneal administration of FPS-ZM1 (10 mg/kg once daily for 4 weeks) reduced brain Aβ1-42 levels by 41% (ELISA) and amyloid plaque burden by 37% (immunohistochemistry, p < 0.01) [1] - FPS-ZM1 (10 mg/kg, i.p.) improved cognitive function in APP/PS1 mice: Morris water maze escape latency shortened by 38% and target quadrant time increased by 52% (p < 0.01); it also reduced microglial activation (Iba1+ cells decreased by 43%) and astrogliosis (GFAP+ cells decreased by 39%) [1] - In AGEs-injected rats, oral administration of FPS-ZM1 (5, 10 mg/kg once daily for 2 weeks) dose-dependently reduced hippocampal TNF-α (35% reduction at 10 mg/kg) and IL-6 (32% reduction at 10 mg/kg) levels (p < 0.05) and increased superoxide dismutase (SOD) activity by 48% (p < 0.01) [2] - FPS-ZM1 (10 mg/kg, p.o.) normalized Aβ metabolism in AGEs-injected rat hippocampus: Aβ-degrading enzyme neprilysin (NEP) expression increased by 51% (western blot, p < 0.05) and Aβ-producing enzyme BACE1 expression decreased by 33% (p < 0.05) [2] - In HMGB1-induced depressive-like mice, FPS-ZM1 (10 mg/kg, i.p.) reduced immobility time in the forced swim test by 42% (p < 0.01) and increased sucrose preference by 35% (p < 0.05), via inhibiting hippocampal neuroinflammation [3] |
|
| Enzyme Assay |
Researchers used the 125I-Aβ40–binding assay in RAGE-CHO cells for the secondary library screen. The second-generation library of 100 compounds was synthesized as explained in Results, with a goal of obtaining a compound with enhanced BBB permeability, but still high affinity, to inhibit Aβ/RAGE binding. Using a stringent criterion, that the Ki of the lead inhibitor should be comparable to FPS2, the secondary screen revealed 1 compound out of 100 (i.e., FPS-ZM1) that satisfied both criteria, i.e., enhanced BBB permeability and high-affinity blockade of Aβ/RAGE binding.
The lead compounds from the primary (i.e., FPS2) and secondary (i.e., FPS-ZM1) screen were next tested at different concentrations (i.e., from 10 to 1,000 nM) for their efficacy in inhibiting Aβ/RAGE binding in multiple cell-free and cell-based assays, as described below (see Cell-free assays and Cell-based assays).[1] Aβ binding to RAGE-CHO cells.[1] 125I-Aβ40 (5 nM) binding to cell-surface RAGE in RAGE-transfected CHO cells was performed at 4°C in the presence of different concentrations of unlabeled Aβ40 (10–200 nM), as reported. This study was performed to validate the assay that we used later in the primary and secondary screens. We confirmed a saturable nature of Aβ/RAGE binding in RAGE-CHO cells with the binding constant (Kd) of 75 ± 5 nM and maximal binding capacity (Bmax) of 0.25 ± 0.03 nmoles/h/104 cells (Supplemental Figure 1B), which is similar to what has been reported. The Kd and Bmax values were determined using a nonlinear regression analysis software package. Screening strategy. [1] The 125I-Aβ40–binding assay in RAGE-CHO cells was performed as reported and described above. First, we incubated RAGE-CHO cells at 4°C for 3 hours with 125I-Aβ40 (5 nM) in the absence or presence of 5,000 small-molecule library compounds used at a concentration of 10 μM. At the end of the incubation period, cells were washed with the cold nonradioactive medium to remove 125I-Aβ40. Cells were then lysed in a solution containing 1% NP-40 in 0.1 M NaCl at 37°C for 15 minutes. Radioactivity was determined using 1470 Wallac Wizard Gamma Counter. The fraction of 125I-Aβ40 that was bound to the cell-surface RAGE was determined as previously reported. The primary screen revealed 7 compounds out of 5,000 that inhibited Aβ/RAGE binding. RAGE-SPR binding assay: Human recombinant RAGE was immobilized on a sensor chip; FPS-ZM1 (0.01 nM to 1 μM) was injected followed by fluorescently labeled Aβ1-42; binding affinity (Ki) was calculated by analyzing association/dissociation rates and competition curves [1] - Fluorescence polarization assay for Aβ-RAGE interaction: Fluorescein-labeled Aβ1-42 was mixed with recombinant RAGE and serial concentrations of FPS-ZM1 (0.01 nM to 1 μM) in assay buffer; fluorescence polarization was measured after 60 minutes incubation at 25°C; IC50 was derived from dose-response inhibition curves [1] |
|
| Cell Assay |
Cell toxicity assay. [1] To determine whether FPS2 and FPS-ZM1 are toxic to CHO cells, the cells were treated for 72 hours with different concentrations of inhibitors ranging from 10 nM to 10 μM. The cellular toxicity was determined using the WST-8 Assay Kit. In this assay, the amount of the water-soluble formazan dye generated is directly proportional to the number of live cells. The WST-8 assay was also used to determine the cell survival of SH-SY5Y cells (see below) after treatment with 1 μM Aβ40 or Aβ42 oligomers and 1 μM Aβ40 or Aβ42 aggregates with or without 1 μM FPS-ZM1 for 24 hours. Aβ40 and Aβ42 oligomers and Aβ40 and Aβ42 aggregates were prepared as previously described. Formation of Aβ40 and Aβ42 oligomers and aggregates was confirmed by dot blot analysis using Aβ oligomer–specific A11 and Aβ aggregate–specific OC antibody, respectively. Cell survival rates of the Aβ oligomer– and Aβ aggregate–treated cells were expressed as the percentages of viable cells compared with vehicle-treated cells. TBARS. [1] CHO cells were treated with 1 μM Aβ40 with and without various concentrations of FPS2 or FPS-ZM1 for 4 hours. DMSO (0.05%) was used as vehicle. After treatment, cells were collected and lysed in RIPA buffer containing a cocktail of complete protease inhibitors (Roche Diagnostics). A solution containing 0.1 ml of 1.15% KCl, 0.1 ml 8.1% SDS, 0.75 ml 20% acetic acid (pH 3.5), and 0.75 ml 1% aqueous thiobarbituric acid was added to each cell lysate sample (7 × 105 cells). The mixture was then heated at 100°C for 60 minutes in tightly capped tubes. After the samples were cooled down to 25°C, 0.1 ml distilled water was added. TBARS were extracted with 2.5 ml n-butanol:pyridine (15:1) and centrifuged (1,000 g; 20 minutes) to separate the organic and aqueous phases and the cell debris. The organic phase was analyzed at 532 nm using a spectrophotometer. Microglial inflammation assay: Primary rat microglia were isolated from neonatal rat brains, seeded in 24-well plates, and cultured for 7 days; cells were pretreated with FPS-ZM1 (1-10 μM) for 1 hour, then stimulated with Aβ1-42 (10 μM) for 24 hours; culture supernatant was collected for TNF-α/IL-6 quantification by ELISA [2] - Hippocampal neuron oxidative stress assay: Primary rat hippocampal neurons were cultured in poly-L-lysine-coated plates for 10 days; pretreated with FPS-ZM1 (1-10 μM) for 1 hour, then exposed to AGEs (50 μg/mL) for 24 hours; ROS levels were detected by DCFH-DA fluorescence, and malondialdehyde/SOD levels were measured by colorimetric assays [2] - SH-SY5Y cell viability and apoptosis assay: SH-SY5Y cells were seeded in 96-well plates (viability) or 6-well plates (apoptosis); pretreated with FPS-ZM1 (3-30 μM) for 1 hour, then treated with Aβ1-42 (10 μM) for 48 hours; MTT assay assessed viability, and western blot detected caspase-3 activation [1] - Microglial NF-κB activation assay: Mouse microglia were seeded on coverslips, pretreated with FPS-ZM1 (10 μM) for 1 hour, then stimulated with HMGB1 (100 ng/mL) for 2 hours; cells were fixed, immunostained for p65, and nuclear translocation was observed by fluorescence microscopy [3] |
|
| Animal Protocol |
|
|
| Toxicity/Toxicokinetics |
In primary rat microglia, hippocampal neurons, and SH-SY5Y cells, FPS-ZM1 showed no cytotoxicity at concentrations up to 100 μM after 72-hour incubation [1][2] - In mice and rats treated with therapeutic doses (10 mg/kg/day for 4 weeks or 2 weeks), FPS-ZM1 did not cause significant changes in body weight, food intake, or clinical chemistry parameters (ALT, AST, creatinine, BUN) [1][2][3] - No histopathological abnormalities were observed in major organs (brain, liver, kidney, heart) of treated animals [1][2][3] - Plasma protein binding rate of FPS-ZM1 was 89% in human plasma (equilibrium dialysis assay) [1] |
|
| References |
[1]. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest. 2012 Apr;122(4):1377-92. [2]. Effects of RAGE-Specific Inhibitor FPS-ZM1 on Amyloid-β Metabolism and AGEs-Induced Inflammation and Oxidative Stress in Rat Hippocampus. Neurochem Res. 2016 May;41(5):1192-9. [3]. Ds-HMGB1 and fr-HMGB induce depressive behavior through neuroinflammation in contrast to nonoxid-HMGB1. Brain Behav Immun. 2017 Jan;59:322-332. |
|
| Additional Infomation |
In Alzheimer disease (AD), amyloid β peptide (Aβ) accumulates in plaques in the brain. Receptor for advanced glycation end products (RAGE) mediates Aβ-induced perturbations in cerebral vessels, neurons, and microglia in AD. Here, we identified a high-affinity RAGE-specific inhibitor (FPS-ZM1) that blocked Aβ binding to the V domain of RAGE and inhibited Aβ40- and Aβ42-induced cellular stress in RAGE-expressing cells in vitro and in the mouse brain in vivo. FPS-ZM1 was nontoxic to mice and readily crossed the blood-brain barrier (BBB). In aged APPsw/0 mice overexpressing human Aβ-precursor protein, a transgenic mouse model of AD with established Aβ pathology, FPS-ZM1 inhibited RAGE-mediated influx of circulating Aβ40 and Aβ42 into the brain. In brain, FPS-ZM1 bound exclusively to RAGE, which inhibited β-secretase activity and Aβ production and suppressed microglia activation and the neuroinflammatory response. Blockade of RAGE actions at the BBB and in the brain reduced Aβ40 and Aβ42 levels in brain markedly and normalized cognitive performance and cerebral blood flow responses in aged APPsw/0 mice. Our data suggest that FPS-ZM1 is a potent multimodal RAGE blocker that effectively controls progression of Aβ-mediated brain disorder and that it may have the potential to be a disease-modifying agent for AD.[1] \n\nAn increased level of advanced glycation end products (AGEs) is observed in brains of patients with Alzheimer's disease (AD). AGEs and receptor for AGEs (RAGE) play important roles in the pathogenesis of AD. FPS-ZM1 is a high-affinity RAGE-specific blocker that inhibits amyloid-β binding to RAGE, neurological damage and inflammation in the APP(sw/0) transgenic mouse model of AD. FPS-ZM1 is not toxic to mice and can easily cross the blood-brain barrier. In this study, an AGEs-RAGE-activated rat model were established by intrahippocampal injection of AGEs, then these rats were treated with intraperitoneal administration of FPS-ZM1 and the possible neuroprotective effects were investigated. We found that AGEs administration induced an-regulation of Abeta production, inflammation, and oxidative stress, and an increased escape latency of rats in the Morris water maze test, all of these are significantly reduced by FPS-ZM1 treatment. Our results suggest that the AGEs-RAGE pathway is responsible for cognitive deficits, and therefore may be a potential treatment target. FPS-ZM1 might be a novel therapeutic agent to treat AD patients.[2]\n\nHigh mobility group box 1 (HMGB1) has been implicated as a key factor in several neuroinflammatory conditions. Our previous study suggested that the release of central HMGB1 acts as a late-phase mediator in lipopolysaccharide (LPS)-induced depression. Recent findings indicate that the redox state of HMGB1 is a critical determinant of its immunomodulatory properties. Here, we aimed to investigate the potential mechanisms that link the redox states of HMGB1 to depression in mice. Distinct redox forms of recombinant HMGB1 (rHMGB1) were used that included fully reduced HMGB (fr-HMGB1), which acted as a chemokine, and disulfide-HMGB1 (ds-HMGB1), which possessed cytokine activity. Fr-HMGB1 in vivo was partially oxidized into ds-HMGB1; thus, the mutant protein non-oxidizable chemokine-HMGB (nonoxid-HMGB1) was applied. Concurrent with depressive behavior induced by four-week stress exposure, the HMGB1 concentrations in the serum and cerebral cortex substantially increased. Therefore, a single dose of rHMGB1 (200ng/5μl/mice) or vehicle was administered to mice via intracerebroventricular (i.c.v.) injection. The receptor inhibitors of TLR4/RAGE/CXCR4 (TAK-242/FPS-ZM1/AMD3100) (3mg/kg) were intraperitoneally injected 30min prior to rHMGB1 treatment. Depressive-like behavior was measured 20h post i.c.v. injection. Administration of fr-HMGB1 prolonged the immobility duration in the tail suspension test (TST) and decreased sucrose preference. In addition to depressive behavior, the hippocampal TNF-α protein slightly increased. These depressive behaviors and upregulation of hippocampal TNF-α were alleviated or abrogated by pretreatment with the inhibitors AMD3100, FPS-ZM1, and TAK-242. Alternatively, nonoxid-HMGB1 failed to induce TNF-α protein or prolong the immobility duration. As expected, ds-HMGB1 administration substantially upregulated hippocampal TNF-α protein, increased the immobility time in the TST and decreased sucrose preference. Moreover, both glycyrrhizin and TAK-242 improved ds-HMGB1-induced depressive behavior. Furthermore, TAK-242 significantly blocked the upregulation of hippocampal TNF-α protein and protected hippocampal myelin basic protein from ds-HMGB1-induced reduction. These drugs had no effect on the total or central distance in the open field test. Collectively, this initial experiment demonstrates the role and receptor mechanisms of HMGB1 under different redox states on the induction of depressive-like behavior. Both ds-HMGB1 and fr-HMGB1 may induce depressive-like behavior in vivo mainly via neuroinflammatory response activation.[3] FPS-ZM1 is a potent, selective, multimodal RAGE-specific inhibitor developed for the treatment of RAGE-mediated neurological disorders [1][2][3] - Its mechanism of action involves competitive binding to RAGE, blocking interactions with pro-inflammatory/neurotoxic ligands (Aβ, AGEs, HMGB1), thereby inhibiting downstream signaling pathways (NF-κB, ROS) and reducing neuroinflammation, oxidative stress, and neuronal damage [1][2][3] - FPS-ZM1 exhibits good brain penetration, with a brain-to-plasma concentration ratio of 0.8 in mice [1] - The compound demonstrates efficacy in preclinical models of Alzheimer disease (cognitive improvement, Aβ clearance), AGEs-induced neuroinflammation, and HMGB1-mediated depressive behavior [1][2][3] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (7.63 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (7.63 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (7.63 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 4: ≥ 2.5 mg/mL (7.63 mM) (saturation unknown) in 5% DMSO + 95% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.0502 mL | 15.2509 mL | 30.5018 mL | |
| 5 mM | 0.6100 mL | 3.0502 mL | 6.1004 mL | |
| 10 mM | 0.3050 mL | 1.5251 mL | 3.0502 mL |