Physicochemical Properties
| Molecular Formula | C29H43NO2S |
| Molecular Weight | 469.728 |
| Exact Mass | 469.301 |
| Elemental Analysis | C, 74.15; H, 9.23; N, 2.98; O, 6.81; S, 6.83 |
| CAS # | 202340-45-2 |
| PubChem CID | 9804740 |
| Appearance | Light brown to khaki solid powder |
| Vapour Pressure | 4.95E-10mmHg at 25°C |
| LogP | 8.724 |
| Hydrogen Bond Donor Count | 2 |
| Hydrogen Bond Acceptor Count | 3 |
| Rotatable Bond Count | 15 |
| Heavy Atom Count | 33 |
| Complexity | 522 |
| Defined Atom Stereocenter Count | 1 |
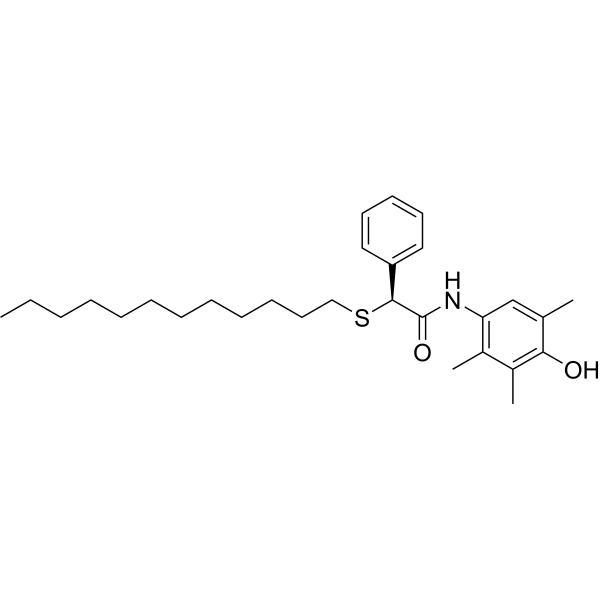
| SMILES | O=C(NC1=CC(C)=C(O)C(C)=C1C)[C@@H](SCCCCCCCCCCCC)C2=CC=CC=C2 |
| InChi Key | ZXEIEKDGPVTZLD-NDEPHWFRSA-N |
| InChi Code | InChI=1S/C29H43NO2S/c1-5-6-7-8-9-10-11-12-13-17-20-33-28(25-18-15-14-16-19-25)29(32)30-26-21-22(2)27(31)24(4)23(26)3/h14-16,18-19,21,28,31H,5-13,17,20H2,1-4H3,(H,30,32)/t28-/m0/s1 |
| Chemical Name | (2S)-2-dodecylsulfanyl-N-(4-hydroxy-2,3,5-trimethylphenyl)-2-phenylacetamide |
| Synonyms | L-0081; Eflucimibe; 202340-45-2; Eflucimibe [INN]; EFLUCIMIBUM; UNII-3DK1X2C37M; eflucimiba; 3DK1X2C37M; f12511; F-12511; Eflucimibe |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | ACAT/acyl-CoA-cholesterol acyltransferase |
| ln Vitro |
New Method:
We report a simple method to encapsulate ACAT inhibitors, using the potent hydrophobic ACAT inhibitor F12511 as an example. By mixing DSPE-PEG2000, egg phosphatidylcholine (PC), and F12511 in ethanol, followed by drying, resuspension and sonication in buffer, we show that F12511 can be encapsulated as stealth liposomes at high concentration.
Results: We successfully incorporated F12511 into nanoparticles and found that increasing PC in the nanoparticles markedly increased the amount of F12511 incorporated in stealth liposomes. The nanoparticles containing F12511 (Nanoparticle F) exhibit average size of approximately 200nm and are stable at 4°C for at least 6 months. Nanoparticle F is very effective at inhibiting ACAT in human and mouse neuronal and microglial cell lines. Toxicity tests using mouse primary neuronal cells show that F12511 alone or Nanoparticle F added at concentrations from 2–10μM for 24-, 48-, and 72-hours produces minimal, if any, toxicity [2]. |
| ln Vivo |
F12511 Is Present in the Brain after Single IV Injection of Nanoparticle F [1] In order to determine an optimal formulation to inhibit ACAT in the brain, we encapsulated F12511 or K604 as part of a DSPE-PEG2000-based nanoparticle (also called a “stealth liposome”; when delivered to the blood, DSPE-PEG2000 protects the liposome from rapid hepatic degradation in vivo). Nanoparticles were prepared based on the procedure described before. We previously found that when the high-affinity ACAT inhibitor such as F12511 or K604 are added to cells, the inhibition of the ACAT enzyme by F12511 or K604 remains unaffected by cell washing or the cell homogenization preparation process. This finding implicates that once the inhibitor binds to the ACAT enzyme, it stays firmly bound for a few hours before dissociating from the enzyme. Based on this finding, we treated the animals with the inhibitor by IV injection, perfused the mice after the animals were treated (to remove blood contamination from the tissues), then prepared the tissue homogenates, and monitored the ACAT enzyme activity in these tissues by using the mixed micelles enzyme assay described previously (the mixed micelles assay measures the ACAT enzyme activity independent of endogenous lipid composition because it reconstitutes the ACAT protein in mixed micelles with a defined lipid composition). We injected nanoparticles with F12511 (nanoparticle F) at 5.8 mg/kg or with K604 at 40 mg/kg by IV. Four hours after IV, we sacrificed the mice and compared the ACAT enzyme activity remaining in various tissues in nanoparticle-F- or K604-treated mice vs. that of the untreated mice. The results (Figure 1) showed that after using our nanoparticle system, F12511 inhibited the ACAT enzyme in the brain, as well as in the adrenal glands, while K604 only inhibited the ACAT enzyme in the adrenal glands and not in the brain. This result shows that as part of nanoparticles, F12511, but not K604, is permeable to the mouse brain. This result is consistent with the work of Shibuya et al., who showed that K604, a quite hydrophilic molecule, is impermeable to the brain. This result also shows that even at a relatively low dose (5.8 mg/kg), 8 h after IV injection, F12511 is still able to inhibit ACAT activity in the brain by more than 50%. Overall, our results demonstrate that, in our nanoparticle system, F12511 exhibited significantly stronger inhibitory effects on ACAT activities in the mouse brain compared to K604. Therefore, the subsequent focus of our study will be on understanding the pharmacokinetics, biodistribution, and efficacy of nanoparticle F in the brain. To determine the extent to whether F12511 alone reaches the brain parenchyma to inhibit ACAT, we conducted a study involving the daily oral gavage administration of nanoparticle F for four consecutive days, and we measured the ACAT activity 6 h after the final administration. Due to the hydrophobic nature of F12511, its delivery in animals at a high dose poses challenges. Previous studies have utilized a delivery vehicle, such as cyclodextrin, to deliver F12511 in animals. In this experiment, we employed an oral gavage administration of the nanoparticle F (Figure S1) as a means of ensuring the high-dose delivery of F12511. It is important to note that liposome-based nanoparticles are expected to degrade in the intestine upon oral delivery. Consequently, F12511 is released into the bloodstream, mimicking the administration of F12511 alone. Our result (Figure S1) revealed a lack of ACAT activity inhibition in the brain even after four daily administrations. However, significant ACAT activity inhibition was observed in peripheral tissues such as the adrenal glands and livers. These findings suggested that without a carrier, the penetration of F12511 into the brain parenchyma is insufficient to inhibit ACAT activity. To examine if IV injection of nanoparticle F causes the preferential accumulation of F12511 in adrenals or in any other tissues, we conducted mass analyses of nanoparticle F by using a LC/MS/MS-based procedure described. The results (Figure 2, top) showed that after a single IV injection of nanoparticle F at a high dose (46 mg F12511/kg), at 4 h, the F12511 content in the plasma reached values close to 30 µM. To compare this value with the values in the literature, it was previously known that F12511 can be solubilized at a high concentration by cyclodextrin. When mice were orally fed with an F12511/cyclodextrin complex, the maximal blood F12511 concentration that could be reached was only 0.75 µM (data retrieved from the Pierre Fabre patent US patent 5990173); i.e., a value 30-fold less than the method described here can achieve. The results in the bottom graph of Figure 2 showed that after a single IV of nanoparticle F, the F12511 content decreased rapidly in the plasma, adrenals, livers, and brains with time (from 4 to 24 h); these results, which were collected via mass spectrometry analyses, indicated that there was no evidence for the preferential accumulation of F12511 in the adrenals. We also monitored the ACAT enzyme activities in various tissues 4 to 48 h after a single IV injection of nanoparticle F at 46 mg/kg. The results (Figure 3) showed that similar to the F12511 tissue content analyses results (Figure 2, bottom), the ACAT enzyme activity in various tissues (including the adrenal glands) steadily returned to near-normal levels within 24 to 48 h, demonstrating that there is no evidence to suggest the prolonged, preferential accumulation of F12511 in the adrenals. Nanoparticle F Attenuates Neuroinflammation in the Aging 3xTg AD Mice [1] The aging 3xTg AD mice exhibited chronic neuroinflammation. Here we tested if nanoparticle F may attenuate neuroinflammation in these mice at 16–20 months of age. After a two-week IV injection to these mice, we prepared whole brain homogenates and analyzed 31 different mouse cytokines by using Milliplex® technology. The result in Figure 8A compares the average cytokine expression level between the different experimental treatment groups. Our data suggested that, when compared to the nanoparticle treatment alone, the treatment with nanoparticle F resulted in a decrease in the trend of 27 out of the 31 analyzed cytokines, of which most were proinflammatory in nature (first two columns from the left, Figure 8A). In some cytokines, such as TNF-α and Eotaxin, the nanoparticles and F12511 worked additively to reduce their levels (Figure 8A). To further examine the effect of the nanoparticles and nanoparticle F in the aging 3xTg AD mice, we plotted individual cytokines data that were previously shown in the literature to be related to neuroinflammation and Alzheimer’s disease, including IL-1α, IL-1β, IL-6, TNF-α, IL12-p40, Eotaxin, MIP-1a, and MCP1 (Figure 8B)). For the cytokines such as IL-1α, IL-1β, IL-6, IL-12p40, MIP-1a, and MCP1, the treatment with nanoparticle F reduced their level compared with the treatment with nanoparticles alone, where a minimal effect from the nanoparticles alone was observed when compared to the untreated 3xTg AD mice (Figure 8B). In cytokines such as TNF-α and Eotaxin, the nanoparticles-alone treatment reduced the cytokines level compared to the untreated 3xTg AD mice (Figure 8B). The treatment with nanoparticle F further reduced the cytokines level compared to the nanoparticles-alone treatment, highlighting the synergetic effect of the nanoparticles and F12511 (Figure 8B). We used Volcano plots to illustrate the difference between all 31 cytokine expressions between each treatment group (shown in Figure S2). We note that the effects of the nanoparticles and nanoparticle F on the cytokine expression were cytokine specific. Together, these results suggested that the treatment with nanoparticles alone and nanoparticle F reduced most proinflammatory cytokine expressions in the aging 3xTg AD mice; human Tau and HPTau were previously shown to be highly inflammatory in the brain, and the removal of these aggregated protein species could restore proper brain function by ameliorating neuroinflammation. We suspect that the effect of the nanoparticles and nanoparticle F treatment on inflammatory cytokines could have been in part due to the ability of the nanoparticles and nanoparticle F treatment to reduce the Aβ, human Tau, and HPTau levels. |
| Enzyme Assay |
Measuring ACAT Activity by Mixed Micelle [1] WT C57BL/6 mouse tissues (forebrain, cerebellum/brain stem, adrenal glands, and liver) were homogenized by using the Next Advance Bullet Blender homogenizer with stainless steel beads; they were mixed twice at a scale of 8, with each mixing session lasting 3 min, while maintaining a temp at 4 °C. The buffer used contained 2.5% 3-((3-Cholamidopropyl) dimethylammonio)-1-propanesulfonate (CHAPS) and 1 M KCl in 50 mM Tris at pH 7.8. Aliquots of the tissue homogenates were then transferred to prechilled glass tubes that contained the mixed liposomal mixture of 9.3 mM taurocholate, 10.8 mM phosphatidylcholine (egg yolk), and 1.8 mM cholesterol. The tubes were vortexed and kept on ice. The samples were then incubated in a 37 °C shaking water bath with 10 nmole 3H-oleoyl CoA/BSA added to start the enzyme reaction for 10 min. The assay was stopped by adding CHCl3:MeOH (2:1), vortexed, and centrifuged at 500 rpm for 10 min. The top phase was removed, and the bottom phase was blow dried by N2, and 100 µL of ethyl acetate was added to each tube, vortexed, and spotted on a thin-layer chromatography (TLC) plate. The TLC solvent system used was petroleum ether:ethyl ether:acetic acid (90:10:1). The cholesteryl ester band with an Rf value at 0.9 was visualized by iodine staining, scraped from the plate, and measured by a scintillation counter for radioactivity. In vitro mixed liposomal ACAT activity assay: [2] This method was done as described in Chang et al., 1998 and Neumann et al., 2019. Monolayers of cells were treated for 4 hours with either control (dimethyl sulfoxide (DMSO)), or with 0.5μM F12511 in DMSO. Cells were then washed with drug-free media kept at 37°C for various amounts of time. At the indicated times, cells were washed with cold PBS, then lysed in a 2.5% 3-((3-Cholamidopropyl) dimethylammonio)-1-propanesulfonate (CHAPS), 1M KCl in 50mM Tris at pH 7.8 buffer. Lysed cells were collected and aliquoted into pre-chilled tubes containing the mixed liposomal mixture of taurocholate, phosphatidylcholine, and cholesterol. Tubes were vortexed and kept on ice for 5 minutes. Samples were then incubated in a shaking 37°C water bath with 3H-oleoyl CoA added for 10 minutes. The assay was stopped by adding CHCl3:MeOH (2:1). Lipids were extracted and analyzed by the same TLC method described under “2.4.1. Intact cell cholesteryl esterification by 3H-Oleate pulse.” |
| Cell Assay |
Wide-field fluorescence and differential interference contrast (DIC) microscopy: [2] Mouse and human neuronal (N2a and SH-SY5Y) and microglial (N9 and HMC3) cells were imaged using the 60x objective of Nikon Eclipse Ti-E microscope. Cells were plated in Mattek 12-well glass bottom plates (1.5 glass thickness, 14mm glass diameter, uncoated) for live-cell imaging. Prior to plating, the plates were coated with poly-d-lysine overnight, washed 3 times with sterile water, and washed once with PBS. The evening before imaging, cell media (described under “2.2. Cell culture”) was changed. On the day of imaging, cells were treated for 2 hours with 0.5μM EtOH (control), F12511 (EtOH), Nanoparticle F, or DSPE-PEG2000/PC. The cells were then stained for nuclear visualization with membrane-permeable Hoechst 33258 solution (Sigma-Aldrich, Catalog #94403) for 20–30 minutes and washed 3 times with PBS. Cells were imaged live in phenol-free media (described under “2.2. Cell culture”) at 37°C with 5% CO2 using Tokai Hit stage top incubation system. Toxicity experiments [2] Lactate dehydrogenase (LDH) toxicity assay: [2] Primary cortical neurons were harvested and maintained as described under “2.2. Cell culture.” Neurons were treated with either F12511 alone (dissolved in EtOH), F12511/DSPE-PEG2000/PC nanoparticles (Nanoparticle F), or DSPE-PEG2000/PC nanoparticles at 2–100μM concentrations for 24-, 48-, or 72-hours. After treatment, the conditioned media from the cells was collected, spun at 12,000rpm for 30 minutes to remove cellular debris, supernatant transferred to new tube, and frozen at −80°C until time of assay. Using Cytotoxicity Detection Kit (LDH), media and reaction mixture were mixed, incubated for 30 minutes at room temperature protected from light, and the absorbance was measured at 490nm. Intact cell cholesteryl esterification by 3H-Oleate pulse: [2] This procedure was according to procedure described in Chang et al., 1986. Monolayers of cells were treated for 2 hours with various concentrations of either control (ethanol or PBS), DSPE-PEG2000/PC nanoparticles, F12511 or K-604 (dissolved in ethanol), or F12511/DSPE-PEG2000/PC (Nanoparticle F) or K-604/DSPE-PEG2000/PC (Nanoparticle K) nanoparticles. Following treatment, cells were pulsed at 37°C for 1–3 hours with 3H-oleate/fatty acid free BSA, then washed with cold PBS, lysed by adding cold 0.2M NaOH at appropriate volume, and placed on orbital shaker for 40 minutes. The solubilized cell slurries were collected into glass tubes, neutralized with 3M HCl and 1M KH2PO4, and lipids were extracted with CHCl3:MeOH (2:1) and water. Samples were vortexed and centrifuged at 500rpm for 10 minutes. The top-phase was removed, and the bottom phase was blown dried by N2 using N-evap apparatus. Dried samples were vortexed with ethyl acetate and spotted on TLC plate. The solvent system used was petroleum ether:ethyl ether:acetic acid (90:10:1). The cholesteryl ester band was scraped from the TLC and measured by scintillation counter for radioactivity. |
| Animal Protocol |
WT C57BL/6 mice were either left untreated, treated with control DSPE-PEG2000/PC nanoparticles, or nanoparticles that contained F12511 (Nanoparticle F). The control and nanoparticle-F-treated mice received 7 daily IV injections and were sacrificed 48 h after the last injection. The mice were anesthetized with Avertin (2% in PBS) and, after confirming by a lack of a toe-pinch reflex, were slowly perfused with 10 mL of 4% sucrose in PBS followed by 10 mL of 4% paraformaldehyde (PFA) in a 4% sucrose solution in PBS. The tissues were collected and kept in PFA at 4 °C on a sample mini rotator overnight before switching the solution to 4% sucrose in the PBS. The tissues were then collected into histology cassettes, embedded into paraffin blocks, and sectioned onto slides with a hematoxylin and eosin (H&E) stain [1].
F12511 was quantified in mouse tissues via LC-MS/MS and CP113,818 (a research gift from Pfizer), and a closely related ACAT inhibitor was used as the internal standard. Both compounds were dissolved in DMSO at 10 mg/mL and stored at −40 °C. Subsequent working dilutions were made in fresh acetonitrile (ACN) daily. Calibrators and quality control measures were used in the appropriate matrix: C57BL/6 plasma (anticoagulant: K3-EDTA, Innovative Research), brain homogenate, liver homogenate, and adrenal gland homogenate. The tissues were homogenized at 0.1 g/mL in diH2O by using stainless steel beads and a Next Advance Bullet Blender. All the samples (50 µL) were protein precipitated, with 150 µL of 10 ng/mL CP113,818 in ACN acting as an internal standard and added via vortex for 1 min and centrifugation for 5 min at 15,000 rpm. In total, 150 µL of supernatant was transferred to amber autosampler vials, and 10 µL was injected on the LC-MS/MS system. HPLC separation was achieved with isocratic conditions of 5% diH2O, 95% methanol, and 0.1% formic acid over 2.5 min at a flow rate of 1.5 mL/min on a Phenomenex Luna C18 100 × 4.6 mm and 3 micron column fitted with a 10 × 4 mm C18 guard at 40 °C. A TSQ Vantage mass spectrometer was operated in positive ion mode with a collision pressure of 1.3 mTorr to measure F12511 (470.242→268.120 m/z) and CP113,818 (471.177→201.040 m/z) with collision energies of 16 and 21, respectively, and S-Lens values of 139 and 187, respectively. The heated ESI source was operated with a spray voltage of 4500 V, vaporizer temperature at 500 °C, capillary temperature at 250 °C, and sheath and auxiliary gases at 30 and 15 arbitrary units, respectively. The quantitative range for F12511 was 0.3–1000 ng/mL for the samples from the liver, adrenal glands, brain, and whole blood, and 0.5–1000 ng/mL for the samples from the plasma [1]. Oral Gavage to Mice: The gavage tube was filled with the volume equivalent to 10 mL/kg of the mouse weight, with either the nanoparticles alone or nanoparticle F at the dose of 46 mg F12511/kg. The mice were restrained by tightly scruffing them with one hand. The gavage tube was then placed in the diastema of their mouth and then advanced along the upper palate until the esophagus was reached. The plunger was slowly and steadily depressed to administer the solution into the stomach. After the injection, the gavage needle was removed and the mouse was placed in a clean and comfortable environment to recover and to be monitored [1]. |
| ADME/Pharmacokinetics |
Cholesterol is essential for cellular function and is stored as cholesteryl esters (CEs). CEs biosynthesis is catalyzed by the enzymes acyl-CoA:cholesterol acyltransferase 1 and 2 (ACAT1 and ACAT2), with ACAT1 being the primary isoenzyme in most cells in humans. In Alzheimer’s Disease, CEs accumulate in vulnerable brain regions. Therefore, ACATs may be promising targets for treating AD. F12511 is a high-affinity ACAT1 inhibitor that has passed phase 1 safety tests for antiatherosclerosis. Previously, we developed a nanoparticle system to encapsulate a large concentration of F12511 into a stealth liposome (DSPE-PEG2000 with phosphatidylcholine). Here, we injected the nanoparticle encapsulated F12511 (nanoparticle F) intravenously (IV) in wild-type mice and performed an HPLC/MS/MS analysis and ACAT enzyme activity measurement. The results demonstrated that F12511 was present within the mouse brain after a single IV but did not overaccumulate in the brain or other tissues after repeated IVs. A histological examination showed that F12511 did not cause overt neurological or systemic toxicity. We then showed that a 2-week IV delivery of nanoparticle F to aging 3xTg AD mice ameliorated amyloidopathy, reduced hyperphosphorylated tau and nonphosphorylated tau, and reduced neuroinflammation. This work lays the foundation for nanoparticle F to be used as a possible therapy for AD and other neurodegenerative diseases. [1] To compare the HPLC/MS/MS analyses of F12511, the result showed that at the 12 to 24 h time point, about 0.5% to 1% of F12511 in the plasma was found in the brain interior (Figure 2, table on top). Together, we speculate that soon after the IV injection, a portion of F12511 may dissociate from the nanoparticle and undergo rapid degradation by the liver, while other portions of F12511 might enter the brain along with the nanoparticle. [1] |
| References |
[1]. Stealth Liposomes Encapsulating a Potent ACAT1/SOAT1 Inhibitor F12511: Pharmacokinetic, Biodistribution, and Toxicity Studies in Wild-Type Mice and Efficacy Studies in Triple Transgenic Alzheimer’s Disease Mice. Int J Mol Sci. 2023 Jul 2;24(13):11013. [2]. Facile method to incorporate high-affinity ACAT/SOAT1 inhibitor F12511 into stealth liposome-based nanoparticle and demonstration of its efficacy in blocking cholesteryl ester biosynthesis without overt toxicity in neuronal cell culture. J Neurosci Methods. 2021 Dec 7;367:109437. [3]. Analysis of eflucimibe and related impurities by highly sensitive liquid chromatography-electrospray-tandem mass spectrometry. The Journal of Separation Science. 2003, 26, 15-16, 1353-1358. |
| Additional Infomation |
Eflucimibe is a member of acetamides. Eflucimibe is a dodecylthio-phenylacetanilide derivative with hypocholesterolemic and anti-atherosclerotic properties. Eflucimibe is an inhibitor of acyl-Coenzyme A:cholesterol acyltransferase (ACAT), an intracellular enzyme which catalyzes the esterification of cholesterol into cholesteryl esters and is believed to be involved in intestinal cholesterol absorption, lipoprotein assembly in the liver, steroid hormone production in the adrenal cortex, and foam cell formation in atherosclerotic lesions. Eflucimibe (F12511) is a new and promising acyl-coenzyme A cholesterol O-acyl-transferase (ACAT) inhibitor, developed at the Pierre Fabre Research Center for the treatment of atherosclerosis. Owing to the synthetic pathway, residual impurities may be present (at very low levels) in the final product, which is currently analysed by high-performance liquid chromatography (HPLC) with UV detection. In this work, a faster, specific, and sensitive analytical method has been developed for the determination of eflucimibe and its four related impurities. Electrospray-tandem mass spectrometry (ESI-MS/MS) coupled with HPLC is demonstrated to efficiently enhance the detection limits (at least 1200-fold), within a 4-minute run time. After optimisation of the ESI and MS/MS parameters for each of the five compounds, quantitative analysis was performed in the multiple reaction monitoring (MRM) mode and the calibration curves yielded correlation coefficients (r ) greater than 0.9996. Finally, this method was successfully applied to the assay of a real batch sample.[3] Using ACAT inhibitors for AD drug treatment has been of interest for two decades. Since essentially all the ACAT inhibitors are quite hydrophobic, methods to solubilize the ACAT inhibitors and to administer these inhibitors in vivo are key hurdles. The method for delivering the ACAT inhibitors CP-113,818 and CI-1011 in mouse models for AD (Hutter-Paier et al., 2004; Huttunen et al., 2010) was done through the use of a slow injection method that involved a custom made slow-release pellet; the composition of the pellet remained proprietary. A different method to deliver ACAT1 specific inhibitor K-604 intranasally was recently reported (Shibuya et al., 2019). Here, Shibuya and colleagues found that at acidic pH (pH 2.3), K-604 became quite water soluble, and when injected intranasally as a solution at pH 2.3, it significantly inhibited cholesteryl ester content in the mouse brain. However, the long-term safety of this method is unknown. In other reports, Bougáret and colleagues found that F12511 can be solubilized at high concentration by forming a complex with cyclodextrin (Bougáret et al., 2005) and Lee and colleagues reported that CI-1011 can be solubilized in human albumen at high concentration (Lee et al., 2015). While these methods look promising, they do not offer the option for nanoparticle tagging. Here we report a simple and efficient method to encapsulate a high-affinity ACAT inhibitor F12511 in DSPE-PEG2000/PC nanoparticles. With the addition of PC, the encapsulation of F12511 increased nearly 6-fold, allowing a very high concentration of F12511 to be incorporated. Nanoparticle F exhibits average size of 200nm and is stable at 4°C for several months. Importantly, efficacy tests show that Nanoparticle F is very effective in inhibiting ACAT enzyme activity in human and mouse neuronal and microglial cell lines, and neither F12511 nor Nanoparticle F appear to exhibit detectable toxicity when given to mouse primary neuronal cells at concentrations ranging from 2–10μM for up to 3 days. The main advantage for this nanoparticle system is its potential impact for in vivo delivery. While F12511 alone (dissolved in DMSO or EtOH) is more potent at inhibiting ACAT in cell culture than Nanoparticle F (as seen in Figures 3 and 4), this would not be feasible for in vivo work. F12511 is very hydrophobic and must be in an emulsion, complexed to something, or as discussed here, encapsulated in nanoparticles for in vivo delivery. We are currently working on delivering Nanoparticle F and Nanoparticle K in vivo. Another benefit of this method is that it can be expanded to produce tagged nanoparticles to increase the specificity of targeting tissues. It also offers opportunity to incorporate water soluble agent(s) within the aqueous space of the nanoliposomes for potential combinatorial therapy. One of the key benefits of this method is the ease of reproducing and characterizing the nanoparticles. This new method will give researchers the opportunity to deliver ACAT inhibitors and other hydrophobic compounds/inhibitors at high concentrations as potential therapies to treat various human diseases, including AD. We also anticipate that by using DSPE-PEG2000 and PC based nanoparticles, the bioavailability and half-life of F12511 in vivo will be significantly increased (Allen et al., 1993). We are currently testing the effects of Nanoparticle F and Nanoparticle K in the systemic tissues as well as in the central nervous system (CNS) of mice in vivo. [2] |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1289 mL | 10.6444 mL | 21.2888 mL | |
| 5 mM | 0.4258 mL | 2.1289 mL | 4.2578 mL | |
| 10 mM | 0.2129 mL | 1.0644 mL | 2.1289 mL |