Edotecarin (formerly ED-749; J-107088; J 107088; PF 804950) is a novel and potent inhibitor of topoisomerase I that can induces single-strand DNA cleavage with IC50 of 50 nM. The novel indolocarbazole edotecarin differs from other topoisomerase I inhibitors both pharmacokinetically and pharmacodynamically. In vitro, it is more potent than camptothecins and has a variable cytotoxic activity in 31 different human cancer cell lines. Edotecarin also possesses greater than additive inhibitory effects on cell proliferation when used in combination with other agents tested in vitro against various cancer cell lines. The present in vivo studies were done to extend the in vitro findings to characterize the antitumor effects of edotecarin when used either alone or in combination with other agents (i.e., 5-fluorouracil, irinotecan, cisplatin, oxaliplatin, and SU11248) in the HCT-116 human colon cancer xenograft model. Treatment effects were based on the delay in onset of an exponential growth of tumors in drug-treated versus vehicle control-treated groups. In all studies, edotecarin was active both as a single agent and in combination with other agents. Combination therapy resulted in greater than additive effects, the extent of which depended on the specific dosage regimen. Toxicity in these experiments was minimal. Of all 359 treated mice, the six that died of toxicity were in the high-dose edotecarin/oxaliplatin group. The results suggest that edotecarin may serve as effective chemotherapy of colon cancer when used as a single agent, in combination with standard regimens and other topoisomerase inhibitors or with novel agents, such as the multitargeted tyrosine kinase inhibitor SU11248.
Physicochemical Properties
| Molecular Formula | C29H28N4O11 |
| Molecular Weight | 608.55282 |
| Exact Mass | 608.175 |
| CAS # | 174402-32-5 |
| PubChem CID | 9808998 |
| Appearance | Yellow to orange solid powder |
| Density | 1.96g/cm3 |
| Boiling Point | 1047.5ºC at 760 mmHg |
| Flash Point | 587.3ºC |
| Vapour Pressure | 0mmHg at 25°C |
| Index of Refraction | 1.886 |
| LogP | -0.7 |
| Hydrogen Bond Donor Count | 10 |
| Hydrogen Bond Acceptor Count | 12 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 44 |
| Complexity | 1120 |
| Defined Atom Stereocenter Count | 5 |
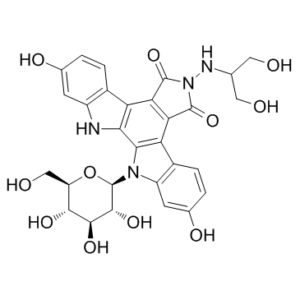
| SMILES | C1=CC2=C(C=C1O)NC3=C4C(=C5C(=C23)C(=O)N(C5=O)NC(CO)CO)C6=C(N4[C@H]7[C@@H]([C@H]([C@@H]([C@H](O7)CO)O)O)O)C=C(C=C6)O |
| InChi Key | QMVPQBFHUJZJCS-NTKFZFFISA-N |
| InChi Code | InChI=1S/C29H28N4O11/c34-7-10(8-35)31-33-27(42)20-18-13-3-1-11(37)5-15(13)30-22(18)23-19(21(20)28(33)43)14-4-2-12(38)6-16(14)32(23)29-26(41)25(40)24(39)17(9-36)44-29/h1-6,10,17,24-26,29-31,34-41H,7-9H2/t17-,24-,25+,26-,29-/m1/s1 |
| Chemical Name | 6-((1,3-dihydroxypropan-2-yl)amino)-2,10-dihydroxy-12-((2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)-12,13-dihydro-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-dione |
| Synonyms | PHA782615; J107088; ED749; PHA-782615; J-107088; ED-749; PHA 782615; J 107088; ED 749. |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| ln Vitro | Edocalin (0.6 μmol/L) enhances DNA-protein complex formation in human colon cancer cells labeled with 3H thymidine in a time-dependent manner [1]. |
| ln Vivo | Edotecarin exhibits potent anti-metastatic properties and can enhance the survival rate of mice with intracranial D-456MG glioma by 83% [1]. Tumor growth was delayed by edocaline for a period of time that varied from 10.45 days at the lowest dose (3 mg/kg) to 24.83 days at the maximum dose (100 mg/kg). When edocalin and irinotecan are used in combination, the anticancer activity in vivo is enhanced as compared to when they are used alone [2]. |
| ADME/Pharmacokinetics |
Metabolism / Metabolites Edotecarin does not form active metabolites and is not a substrate for in vitro P450-mediated metabolism. Biological Half-Life 20 to 25 hours |
| References |
[1]. Edotecarin: a novel topoisomerase I inhibitor. Clin Colorectal Cancer. 2005 May;5(1):27-36. [2]. Antitumor efficacy of edotecarin as a single agent and in combination with chemotherapy agents in a xenograft model. Clin Cancer Res. 2006 May 1;12(9):2856-61. |
| Additional Infomation |
Edotecarin is a novel, non-camptothecin, DNA topoisomerase I inhibitor. It is member of the class of compounds called indolocarbazoles. Edotecarin is a synthetic indolocarbazole with antineoplastic activity. Edotecarin inhibits the enzyme topoisomerase I through stabilization of the DNA-enzyme complex and enhanced single-strand DNA cleavage, resulting in inhibition of DNA replication and decreased tumor cell proliferation. (NCI04) Drug Indication Clinical studies with edotecarin have shown activity in subjects with colorectal cancer, esophageal cancer and other solid tumors. Mechanism of Action Edotecarin inhibits the enzyme topoisomerase I through stabilization of the DNA-enzyme complex and enhanced single-strand DNA cleavage, resulting in inhibition of DNA replication and decreased tumor cell proliferation. Pharmacodynamics Edotecarin, formerly J-107088 is a novel, non-camptothecin, DNA topoisomerase-1 inhibitor. It is part of the class of compounds called indolocarbazoles. It is a novel inhibitor of topoisomerase I that induces single-strand DNA cleavage more effectively than NB-506 or camptothecin (CPT) and at different DNA sequences. The DNA-topoisomerase I complexes induced by edotecarin are more stable than those occurring after exposure to CPT or NB-506. The antitumor activity of edotecarin is less cell cycle dependent than other topoisomerase I inhibitors. Being an indolocarbazole, it is structurally related to staurosporine but does not possess protein kinase inhibitory properties. The antitumor activity of edotecarin has been tested in vitro and in vivo, and inhibition of tumor growth has been observed in breast, cervix, pharynx, lung, prostate, colon, gastric, and hepatic cancer models. Edotecarin is effective on cells that have acquired resistance related to P-glycoprotein. In vitro synergy has been demonstrated when edotecarin was tested in combination with cisplatin, 5-fluorouracil, etoposide, paclitaxel, doxorubicin, vincristine, CPT, and gemcitabine. |
Solubility Data
| Solubility (In Vitro) | May dissolve in DMSO (in most cases), if not, try other solvents such as H2O, Ethanol, or DMF with a minute amount of products to avoid loss of samples |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.6433 mL | 8.2163 mL | 16.4325 mL | |
| 5 mM | 0.3287 mL | 1.6433 mL | 3.2865 mL | |
| 10 mM | 0.1643 mL | 0.8216 mL | 1.6433 mL |