Edicotinib (formerly known as JNJ-527; JNJ-40346527) is a novel selective and orally bioavailable inhibitor of colony-stimulating factor-1 (CSF-1) receptor kinase with anticancer activity. It functions to prevent macrophage survival, proliferation, and differentiation in patients receiving disease-modifying antirheumatic drug (DMARD) therapy but still having active rheumatoid arthritis (RA). Clinical research is being done on this topic. Increased levels of CSF-1 and decreased CD16+ monocytes in JNJ-40346527-treated patients, but not in placebo-treated patients, demonstrated effective target engagement and proof of activity.Pharmacokinetic exposure to JNJ-40346527 and its active metabolites was above the projected concentration needed for pharmacologic activity. A total of 37 patients (58.7%) treated with JNJ-40346527 and 16 (50.0%) treated with placebo reported ≥ 1 adverse event (AE); 1 patient (1.6%) treated with JNJ-40346527 and 3 patients (9.4%) treated with placebo reported ≥ 1 serious AE.
Physicochemical Properties
| Molecular Formula | C27H35N5O2 | |
| Molecular Weight | 461.61 | |
| Exact Mass | 461.279 | |
| Elemental Analysis | C, 70.25; H, 7.64; N, 15.17; O, 6.93 | |
| CAS # | 1142363-52-7 | |
| Related CAS # | 1559069-92-9 (HCl);1142363-52-7; | |
| PubChem CID | 25230468 | |
| Appearance | White to off-white solid powder | |
| Density | 1.2±0.1 g/cm3 | |
| Index of Refraction | 1.591 | |
| LogP | 5.16 | |
| Hydrogen Bond Donor Count | 2 | |
| Hydrogen Bond Acceptor Count | 5 | |
| Rotatable Bond Count | 4 | |
| Heavy Atom Count | 34 | |
| Complexity | 838 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | O1C(C)(C)CC(C2C=CC(=C(C3=CCC(C)(C)CC3)N=2)NC(C2=NC=C(C#N)N2)=O)CC1(C)C |
|
| InChi Key | BNVPFDRNGHMRJS-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C27H35N5O2/c1-25(2)11-9-17(10-12-25)22-21(32-24(33)23-29-16-19(15-28)30-23)8-7-20(31-22)18-13-26(3,4)34-27(5,6)14-18/h7-9,16,18H,10-14H2,1-6H3,(H,29,30)(H,32,33) | |
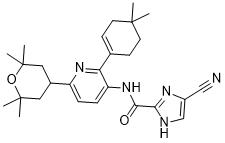
| Chemical Name | 5-cyano-N-[2-(4,4-dimethylcyclohexen-1-yl)-6-(2,2,6,6-tetramethyloxan-4-yl)pyridin-3-yl]-1H-imidazole-2-carboxamide | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
CSF-1R (IC50 = 3.2 nM); KIT (IC50 = 20 nM); FLT3 (IC50 = 190 nM)
Colony-Stimulating Factor 1 Receptor (CSF1R); in vitro IC50 for inhibiting CSF1R phosphorylation in N13 cells: 18.6–22.5 nM; pharmacokinetic/pharmacodynamic (PK/PD)-derived EC50: 196 ng/mL (plasma) and 69 ng/g (brain tissue) [1] - Colony-Stimulating Factor 1 Receptor (CSF1R); no specific IC50, Ki, or EC50 values provided; effective target engagement demonstrated by increased plasma CSF1 levels and decreased CD16+ monocytes in patients [2] |
|
| ln Vitro |
|
|
| ln Vivo |
|
|
| Enzyme Assay |
In vitro assessment of CSF1R phosphorylation [1] The N13 murine microglia cell line (Righi et al., 1991) was cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% foetal bovine serum and 50 U/ml penicillin/streptomycin. Cells were maintained in T75 flasks at 37°C in a 5% CO2 humidified atmosphere. Cells were plated at a density of 2 × 105 cells/cm2 in 6-well plates and cultured overnight to allow adherence. Cells were kept in serum-free medium for 4 h prior to stimulation and then incubated without or with 0.1, 1, 10, 100 or 1000 nM of Edicotinib (JNJ-527;JNJ-40346527) for 30 min. Recombinant CSF1 (100 ng/ml, R&D Systems) was added to respective wells for 5 min, after which cells were immediately lysed in RIPA buffer, supplemented with protease and phosphatase inhibitor cocktails. Protein lysates were concentrated using Microcon-10 kDa Centrifugal Filter Units, according to manufacturer’s instructions and protein concentration was determined using the Pierce BCA Protein Assay Kit. For estimation of IC50, values for CSF1R and ERK1/2 phosphorylation were modelled in a non-linear regression curve using GraphPad prism. The novel selective and orally bioavailable inhibitor of colony-stimulating factor-1 (CSF-1) receptor kinase, edicotinib (previously known as JNJ-40346527), was discovered. CSF1R phosphorylation and ERK1/2 activation assay in N13 cells: N13 cells were seeded and pretreated with serial concentrations of Edicotinib for a set period, then stimulated with CSF1 to activate CSF1R signaling. Cells were lysed in buffer containing protease/phosphatase inhibitors, and total protein was extracted. Protein concentrations were quantified, and equal amounts of protein were separated by SDS-PAGE, transferred to membranes, and probed with primary antibodies against phosphorylated CSF1R, total CSF1R, phosphorylated ERK1/2, and total ERK1/2. Immunoreactive bands were visualized and quantified to assess inhibition efficacy [1] - TSPO autoradiography assay: Brain tissues from ME7 mice (after Edicotinib treatment) were sectioned, and incubated with [3H]-PK11195 (a TSPO ligand) to detect microglial activation. Radioactivity was measured using autoradiography, and signal intensity was quantified to evaluate the effect of Edicotinib on neuroinflammation [1] |
|
| Cell Assay |
Cell Line: N13 microglial cells Concentration: 0.1 nM, 1 nM, 10 nM, 100 nM, 1000 nM Incubation Time: 24 hours Result: Prevented CSF1R and ERK1/2 phosphorylation in N13 microglial cells Microglial proliferation assay (Iba-1/BrdU double immunohistochemistry): Brain sections from ME7 mice (after Edicotinib and BrdU treatment) were deparaffinized, rehydrated, and subjected to antigen retrieval. Sections were incubated with primary antibodies against Iba-1 (microglial marker) and BrdU (proliferation marker), followed by species-matched secondary antibodies (DAB for Iba-1, alkaline phosphatase for BrdU). Proliferating microglia (Iba-1+ BrdU+ cells) were counted per mm² to quantify inhibition [1] - Flow cytometry for microglial quantification: Spinal cord tissues from P301S mice were dissociated into single-cell suspensions, stained with fluorochrome-conjugated antibodies against CD11b and CD45 (microglial markers), and analyzed by flow cytometry. The number of CD11b+ CD45+ cells was quantified to assess Edicotinib-mediated microglial reduction [1] - PCR for cytokine mRNA detection: Total RNA was extracted from spinal cord tissues of P301S mice (after Edicotinib treatment) using a phenol-chloroform method. Complementary DNA (cDNA) was synthesized from total RNA, and quantitative real-time PCR was performed with specific primers for IL1-β and TNFα (pro-inflammatory cytokines) and a reference gene. Relative mRNA expression levels were calculated to evaluate cytokine reduction [1] - Western blot for tau phosphorylation and kinase activation: Spinal cord tissues from P301S mice were lysed, and protein extracts were subjected to SDS-PAGE. Membranes were probed with antibodies against phosphorylated tau (AT8 epitope), phosphorylated JNK, phosphorylated p38, and a loading control. Band intensities were quantified to assess Edicotinib’s effect on tau pathology and kinase activation [1] |
|
| Animal Protocol |
C57BL/6 J (Harlan) mice 3, 10, 30 and 100 mg/kg; 5 days Oral gavage Pharmacological treatments [1] For short-term treatments, Edicotinib (JNJ-527;JNJ-40346527) was dissolved in 0.9% Methocel™ and administered daily (morning) for five consecutive days by oral gavage at doses of 3, 10, 30 and 100 mg/kg. For long term treatments (4–8 weeks), Edicotinib (JNJ-527;JNJ-40346527) was incorporated into mouse chow as previously described by Olmos-Alonso et al. (2016), for a final dose of 30 mg/kg with an average daily ingestion of 5 g of food per mouse. Diet composition was identical in terms of fat, protein, etc. content, with the only addition of the compound. Mouse weight and food consumption were monitored in all experiments, and no differences were found between treated and untreated groups. TSPO autoradiography [1] Mice were terminally anaesthetized with an overdose of sodium pentobarbital and transcardially perfused with 0.9% saline. Brains were harvested, frozen in isopentane at a temperature of −40°C and stored at −80°C. NBH (n = 7), ME7 (n = 8) and ME7 + Edicotinib (JNJ-527;JNJ-40346527) (n = 8) mouse brains were coronally cryosectioned at 20 μm and directly mounted onto glass slides. Slides were incubated at room temperature for 30 min in 100 mM Tris-HCl containing 1 nM [3H]PK11195 (specific activity 82.7 Ci per mmol), washed twice for 6 min in 100 mM Tris-HCl, rinsed dipping into dH2O and air dried. Pharmacokinetics: sample preparation and bioanalytical method [1] Aliquots (10 µl) of plasma and brain homogenate (diluted 1:5 in phosphate buffer) were analysed for Edicotinib (JNJ-527;JNJ-40346527) concentrations using a method based on protein precipitation and HPLC-MS/MS analysis. To each sample, an internal standard (20 µl) and acetonitrile (150 µl) were added. Samples were mixed thoroughly (mechanical shaking for 10 min), and then centrifuged (5000g for 10 min at 4°C). An aliquot of supernatant (20 µl) was dispensed into a LCMS plate and 200 µl of 0.1% formic acid in methanol/water (50:50) were added. Analysis for Edicotinib (JNJ-527;JNJ-40346527) concentrations was performed using HPLC-MS/MS employing positive-ion electrospray ionization (Sciex API 4000) and a Zorbaz Eclipse Phenyl Hexyl, 3.5 μm (50 × 2.1 mm internal diameter) column. Elution was achieved at a flow rate of 0.5 ml/min with isocratic elution of 0.1% formic acid in methanol/water (85:15). The lower limit of quantification was 5–10 ng/ml for plasma and 10 ng/g for brain. The assay was linear up to 4000 ng/ml for plasma and 4000 ng/g for brain. In this randomized, double-blind, placebo-controlled, parallel group study, adults were randomized (2:1) to receive oral Edicotinib (JNJ-527;JNJ-40346527) 100 mg or placebo twice daily through Week 12. Patients with RA had disease activity [≥ 6 swollen/≥ 6 tender joints, C-reactive protein (CRP) ≥ 0.8 mg/dl] despite DMARD therapy for ≥ 6 months. The primary endpoint was change from baseline at Week 12 in the 28-joint Disease Activity Score with CRP (DAS28-CRP). Pharmacokinetic/pharmacodynamic analyses were also performed, and safety was assessed through Week 16.[2] ME7 prion model mouse experiment: Mice were divided into groups: NBH (control), NBH + Edicotinib 30 mg/kg, ME7 (disease control), ME7 + Edicotinib (3, 10, 30, 100 mg/kg; n=6–14 per group). Edicotinib was administered for 5 days, and BrdU was injected 4 times daily during treatment to label proliferating cells. At the end of treatment, plasma and brain tissues were collected to measure Edicotinib concentrations; brain sections were analyzed by immunohistochemistry to quantify microglial proliferation [1] - P301S tauopathy model mouse experiment: P301S transgenic mice and wild-type mice were used; P301S mice were treated with Edicotinib 30 mg/kg for 8 weeks. During treatment, motor function was assessed using the rotarod test. At the end of treatment, spinal cord tissues were collected for flow cytometry (microglial count), PCR (cytokine mRNA), western blot (tau pathology), and Nissl staining (motor neuron count) [1] |
|
| ADME/Pharmacokinetics |
Researchers analysed the concentration of the compound in brain and plasma and found a linear dose dependent increase in JNJ-527 exposure, with an average brain to plasma ratio of 0.65 (Fig. 1E). We then assessed the impact of CSF1R blockade by JNJ-527 on microglial proliferation. Researchers found that JNJ-527 significantly inhibited microglial proliferation (Iba1+ BrdU+ cells) in the hippocampus of ME7 mice from 3 mg/kg, reaching a maximum effect of 80% inhibition at 30 mg/kg (Fig. 1F and G). Based on these data, we generated a sigmoid Emax pharmacokinetic/pharmacodynamics model for inhibition of microglial proliferation and determined that JNJ-527 EC50 was 196 ng/ml or 69 ng/g for plasma and brain exposures, respectively (Fig. 1H). Overall, Researchers demonstrated that JNJ-527 administered at 30 mg/kg significantly blocks microglial proliferation in ME7-prion mice, without altering the dynamics of the population in the healthy brain (Supplementary Fig. 2C and D). Therefore, Researchers used a dose of 30 mg/kg for all subsequent experiments. [1] In ME7 mice, plasma and brain concentrations of Edicotinib showed a linear correlation with administered doses (3, 10, 30, 100 mg/kg), with tissue-to-plasma (T/P) ratios of 0.5–1 [1] - In patients with RA (Phase IIA study), pharmacokinetic exposure to Edicotinib and its active metabolites was confirmed to be above the projected concentration required for pharmacologic activity [2] |
|
| Toxicity/Toxicokinetics |
In patients with RA (Phase IIA study): 58.7% of Edicotinib-treated patients (n=63) reported ≥1 adverse event (AE), compared to 50.0% of placebo-treated patients (n=32); 1.6% of Edicotinib-treated patients reported ≥1 serious AE, vs. 9.4% in the placebo group. No significant changes in hematological parameters, liver, or kidney function were reported (data not explicitly provided) [2] - In ME7 and P301S mouse models, no overt toxicity (e.g., body weight loss, organ damage) was observed at the tested doses (3–100 mg/kg) [1] |
|
| References |
[1]. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice.Brain. 2019 Oct 1;142(10):3243-3264. [2]. Results from a Phase IIA Parallel Group Study of JNJ-40346527, an Oral CSF-1R Inhibitor, in Patients with Active Rheumatoid Arthritis despite Disease-modifying Antirheumatic Drug Therapy.J Rheumatol. 2015 Oct;42(10):1752-60. |
|
| Additional Infomation |
JNJ-40346527 has been used in trials studying the treatment of Health and Arthritis, Rheumatoid. Edicotinib is a small molecule and orally available inhibitor of colony-stimulating factor-1 receptor (CSF1R; FMS) with potential antineoplastic activity. Edicotinib blocks the receptor-ligand interaction between FMS and its ligand CSF1, thereby preventing autophosphorylation of FMS. As a result, unphosphorylated FMS can not activate FMS-mediated signaling pathways, thus potentially inhibiting cell proliferation in FMS-overexpressed tumor cells. FMS, a tyrosine kinase receptor, is overexpressed in certain tumor cell types and plays an essential role in macrophage differentiation, recruitment, and activation as well as the regulation of cell proliferation. \nNeuroinflammation and microglial activation are significant processes in Alzheimer's disease pathology. Recent genome-wide association studies have highlighted multiple immune-related genes in association with Alzheimer's disease, and experimental data have demonstrated microglial proliferation as a significant component of the neuropathology. In this study, we tested the efficacy of the selective CSF1R inhibitor JNJ-40346527 (JNJ-527) in the P301S mouse tauopathy model. We first demonstrated the anti-proliferative effects of JNJ-527 on microglia in the ME7 prion model, and its impact on the inflammatory profile, and provided potential CNS biomarkers for clinical investigation with the compound, including pharmacokinetic/pharmacodynamics and efficacy assessment by TSPO autoradiography and CSF proteomics. Then, we showed for the first time that blockade of microglial proliferation and modification of microglial phenotype leads to an attenuation of tau-induced neurodegeneration and results in functional improvement in P301S mice. Overall, this work strongly supports the potential for inhibition of CSF1R as a target for the treatment of Alzheimer's disease and other tau-mediated neurodegenerative diseases.[1] \n\nObjective: To assess the efficacy and safety of JNJ-40346527, a selective inhibitor of colony-stimulating factor-1 (CSF-1) receptor kinase that acts to inhibit macrophage survival, proliferation, and differentiation in patients with active rheumatoid arthritis (RA) despite disease-modifying antirheumatic drug (DMARD) therapy.\n\nMethods: In this randomized, double-blind, placebo-controlled, parallel group study, adults were randomized (2:1) to receive oral JNJ-40346527 100 mg or placebo twice daily through Week 12. Patients with RA had disease activity [≥ 6 swollen/≥ 6 tender joints, C-reactive protein (CRP) ≥ 0.8 mg/dl] despite DMARD therapy for ≥ 6 months. The primary endpoint was change from baseline at Week 12 in the 28-joint Disease Activity Score with CRP (DAS28-CRP). Pharmacokinetic/pharmacodynamic analyses were also performed, and safety was assessed through Week 16.\n\nResults: Ninety-five patients were treated (63 JNJ-40346527, 32 placebo); 8 patients discontinued treatment (6 JNJ-40346527, 2 placebo) through Week 12. Mean improvements in DAS28-CRP from baseline to Week 12 were 1.15 for the JNJ-40346527 group and 1.42 for the placebo group (p = 0.30); thus, a statistically significant difference was not observed for the primary endpoint. Pharmacokinetic exposure to JNJ-40346527 and its active metabolites was above the projected concentration needed for pharmacologic activity, and effective target engagement and proof of activity were demonstrated by increased levels of CSF-1 and decreased CD16+ monocytes in JNJ-40346527-treated, but not placebo-treated, patients. Thirty-seven (58.7%) JNJ-40346527-treated and 16 (50.0%) placebo-treated patients reported ≥ 1 adverse event (AE); 1 (1.6%) JNJ-40346527-treated and 3 (9.4%) placebo-treated patients reported ≥ 1 serious AE.\n\nConclusion: Although adequate exposure and effective peripheral target engagement were evident, JNJ-40346527 efficacy was not observed in patients with DMARD-refractory active RA. ClinicalTrials.gov identifier: NCT01597739. EudraCT Number: 2011-004529-28.[2] Edicotinib (JNJ-40346527) is a selective CSF1R inhibitor that acts by inhibiting microglial survival, proliferation, and differentiation, and reducing pro-inflammatory cytokine production [1][2] - Preclinically, Edicotinib attenuates tau-induced neurodegeneration and improves functional outcomes in P301S tauopathy mice, supporting its potential for treating Alzheimer’s disease and other tau-mediated neurodegenerative diseases [1] - In a Phase IIA study for active RA (despite DMARD therapy), Edicotinib showed effective peripheral target engagement but no statistically significant efficacy in improving DAS28-CRP (primary endpoint). ClinicalTrials.gov identifier: NCT01597739; EudraCT Number: 2011-004529-28 [2] - In ME7 prion model mice, Edicotinib reduces microglial expansion and restores behavioral alterations (locomotor activity, motor deficits), with potential CNS biomarkers (TSPO autoradiography, CSF proteomics) identified for clinical investigation [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 1.67 mg/mL (3.62 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 16.7 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 1.67 mg/mL (3.62 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 16.7 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. Solubility in Formulation 3: 10 mg/mL (21.66 mM) in 17% Polyethylene glycol 12-hydroxystearate in Saline (add these co-solvents sequentially from left to right, and one by one), suspension solution; with ultrasonication. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1663 mL | 10.8317 mL | 21.6633 mL | |
| 5 mM | 0.4333 mL | 2.1663 mL | 4.3327 mL | |
| 10 mM | 0.2166 mL | 1.0832 mL | 2.1663 mL |