ETP-46321 is a novel, potent and orally bioavailable inhibitor of PI3Kα and PI3Kδ with Kiapp values of 2.3 nM and 14.2 nM, respectively in biochemical assays. PI3K is a crucial target for cancer therapeutics because a variety of human cancers exhibit deregulation of this signaling pathway. In a GEMM mouse tumor model fueled by a K-Ras(G12V) oncogenic mutation, TP-46321 PK/PD studies revealed time-dependent downregulation of AKT(Ser473) phosphorylation, which correlates with compound levels in tumor tissue. ETP-46321 therapy significantly slowed the growth of the tumor.
Physicochemical Properties
| Molecular Formula | C20H27N9O3S | |
| Molecular Weight | 473.55 | |
| Exact Mass | 473.196 | |
| Elemental Analysis | C, 50.73; H, 5.75; N, 26.62; O, 10.14; S, 6.77 | |
| CAS # | 1252594-99-2 | |
| Related CAS # |
|
|
| PubChem CID | 46927938 | |
| Appearance | White to off-white solid powder | |
| LogP | 1.285 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 11 | |
| Rotatable Bond Count | 5 | |
| Heavy Atom Count | 33 | |
| Complexity | 746 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | S(C([H])([H])[H])(N1C([H])([H])C([H])([H])N(C([H])([H])C2=C([H])N3C([H])=C(C4=C([H])N=C(N([H])[H])N=C4[H])N=C(C3=N2)N2C([H])([H])C([H])([H])OC([H])([H])C2([H])[H])C([H])([H])C1([H])[H])(=O)=O |
|
| InChi Key | OHKDVDMWRKFZRB-UHFFFAOYSA-N | |
| InChi Code | InChI=1S/C20H27N9O3S/c1-33(30,31)29-4-2-26(3-5-29)12-16-13-28-14-17(15-10-22-20(21)23-11-15)25-19(18(28)24-16)27-6-8-32-9-7-27/h10-11,13-14H,2-9,12H2,1H3,(H2,21,22,23) | |
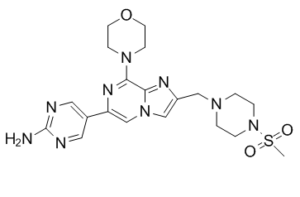
| Chemical Name | 5-[2-[(4-methylsulfonylpiperazin-1-yl)methyl]-8-morpholin-4-ylimidazo[1,2-a]pyrazin-6-yl]pyrimidin-2-amine | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
p110α (Ki = 2.3 nM); PI3Kα-E545K (Ki = 1.77 nM); PI3Kα-E542K (Ki = 1.89 nM); ; p110δ (Ki = 14.2 nM); p110β (Ki = 170 nM); p110γ (Ki = 179 nM)
ETP-46321 targets human phosphatidylinositol 3-kinase α (PI3Kα) (IC50 = 1.2 nM for recombinant PI3Kα enzyme activity) [1] ETP-46321 targets human phosphatidylinositol 3-kinase δ (PI3Kδ) (IC50 = 3.5 nM for recombinant PI3Kδ enzyme activity) [1] ETP-46321 exhibits moderate selectivity over other PI3K isoforms: PI3Kβ IC50 = 89 nM, PI3Kγ IC50 = 126 nM; >100-fold selectivity over class I-like PI3K (PI3Kγ) and class II/III PI3K isoforms (IC50 > 1 μM) [1] |
| ln Vitro |
The PI3K isoform ETP-46321 is chosen for screening. ETP-46321 has a higher Kiapp value (2.3 nM) than isoform. With regard to mTOR and 288 representative kinases, ETP-4632 has been profiled and demonstrated to be a potent PI3K α and δ inhibitor and kinase. ETP-46321 is also tested against three p110 mutant enzymes identified in human cancers (E542K, E545K, and H1047R), and it is equally potent against these mutants as the wild-type protein (Kiapp=2.33, 1.77, and 1.89 nM for PI3Kα-H1047R, PI3Kα-E545K, respectively). In the U2OS cell line, ETP-46321 has an IC50 of 8.3 nM, which inhibits the phosphorylation of AKT. In recombinant PI3K isoform enzyme activity assays, ETP-46321 dose-dependently inhibited PI3Kα (IC50 = 1.2 nM) and PI3Kδ (IC50 = 3.5 nM), with weaker inhibition of PI3Kβ (IC50 = 89 nM) and PI3Kγ (IC50 = 126 nM) [1] - ETP-46321 exhibited potent antiproliferative activity against PI3Kα/δ-dependent human cancer cell lines: IC50 values were 15 nM (MCF-7, breast cancer, PIK3CA mutant), 12 nM (BT474, breast cancer, PIK3CA mutant), 18 nM (Ramos, B-cell lymphoma, PI3Kδ-dependent), 22 nM (SU-DHL-4, diffuse large B-cell lymphoma), and 25 nM (A549, non-small cell lung cancer) after 72-hour treatment (CellTiter-Glo assay) [1] - Western blot analysis in MCF-7 cells showed ETP-46321 (10-50 nM) dose-dependently inhibited PI3K/Akt signaling pathway: 50 nM reduced phosphorylated Akt (p-Akt Ser473) by ~85%, phosphorylated S6 ribosomal protein (p-S6) by ~78%, and phosphorylated GSK3β (p-GSK3β) by ~72% compared to vehicle, with no significant change in total Akt, S6, or GSK3β levels [1] - ETP-46321 (20 nM) induced G1 phase cell cycle arrest in BT474 cells: G1 population increased from 52.3% (vehicle) to 76.5% after 24-hour treatment, accompanied by a decrease in S phase (from 28.6% to 12.8%) and G2/M phase (from 19.1% to 10.7%) (flow cytometry) [1] - ETP-46321 (10-40 nM) dose-dependently induced apoptosis in Ramos cells: 40 nM treatment resulted in an apoptotic rate of 36.8% (Annexin V-FITC/PI staining) compared to 4.2% in vehicle control [1] - ETP-46321 (up to 1 μM) did not affect the viability of normal human mammary epithelial cells (HMEC) or peripheral blood mononuclear cells (PBMCs) (CC50 > 1 μM) [1] |
| ln Vivo |
ETP-46321, is selected for in vivo studies based on its appealing pharmacokinetic profile in BALB-C mice, low in vivo Clearance (0.6 L/h/Kg) and good oral bioavailability (90%). ETP-46321 exhibits a favorable pharmacokinetic profile in mice and is chosen for preliminary in vivo testing in a lung tumor mouse model driven by the K-RasG12V oncogenic mutation. PET imaging techniques show significant tumor growth inhibition and a decrease in tumor metabolic activity[1]. Antitumor efficacy in MCF-7 xenograft model: Female BALB/c nude mice bearing MCF-7 breast cancer xenografts were treated with ETP-46321 (30 mg/kg/day, 60 mg/kg/day, oral) for 21 days. High-dose treatment achieved a tumor growth inhibition (TGI) rate of 75%, reducing tumor weight from 1.35 ± 0.16 g (vehicle) to 0.34 ± 0.06 g. Tumor tissue western blot showed p-Akt Ser473 was reduced by ~78% in high-dose group [1] - Antitumor efficacy in Ramos xenograft model: Female BALB/c nude mice bearing Ramos B-cell lymphoma xenografts were treated with ETP-46321 (20 mg/kg/day, 40 mg/kg/day, oral) for 14 days. High-dose treatment achieved a TGI rate of 72%, reducing tumor weight from 1.12 ± 0.13 g (vehicle) to 0.31 ± 0.05 g [1] - No significant weight loss (vehicle: 22.8 ± 1.2 g vs. high-dose MCF-7 model: 21.7 ± 1.0 g; high-dose Ramos model: 21.5 ± 0.9 g) or overt toxicity (lethargy, organ damage, hematological/biochemical abnormalities) was observed in treated mice [1] |
| Enzyme Assay |
Recombinant PI3K isoform enzyme activity assay: Purified recombinant human PI3Kα, PI3Kδ, PI3Kβ, and PI3Kγ catalytic subunits were incubated with phosphatidylinositol (PI) substrate and ATP (10 μM) in kinase reaction buffer. Serial dilutions of ETP-46321 (0.01-1000 nM) were added, and the mixture was incubated at 30°C for 60 minutes. The reaction was terminated by adding a stop solution, and the amount of phosphorylated PI (PIP) product was quantified using a fluorescent-based detection system. IC50 values were calculated by nonlinear regression of dose-response curves of PIP production inhibition [1] |
| Cell Assay |
Cancer cell antiproliferation assay: MCF-7, BT474, Ramos, SU-DHL-4, and A549 cells were seeded in 96-well plates (5×10³ cells/well) and allowed to attach for 24 hours. Serial dilutions of ETP-46321 (0.1-100 nM) were added, and cells were cultured for 72 hours. Cell viability was measured using a luminescent cell viability assay, and IC50 values were derived from dose-response curves [1] - PI3K/Akt signaling pathway inhibition assay: MCF-7 cells were seeded in 6-well plates (2×10⁵ cells/well) and treated with ETP-46321 (10-50 nM) for 24 hours. Cells were lysed in RIPA buffer, and proteins were separated by SDS-PAGE. Western blot was performed using antibodies against p-Akt Ser473, total Akt, p-S6, total S6, p-GSK3β, total GSK3β, and GAPDH (loading control) [1] - Cell cycle analysis: BT474 cells were seeded in 6-well plates (2×10⁵ cells/well) and treated with ETP-46321 (20 nM) for 24 hours. Cells were harvested, fixed with ethanol, stained with propidium iodide (PI), and analyzed by flow cytometry to determine cell cycle distribution [1] - Apoptosis assay: Ramos cells were seeded in 6-well plates (2×10⁵ cells/well) and treated with ETP-46321 (10-40 nM) for 48 hours. Cells were stained with Annexin V-FITC and PI, then analyzed by flow cytometry to quantify apoptotic rate [1] - Normal cell viability assay: HMEC and PBMCs were seeded in 96-well plates (5×10³ cells/well) and treated with ETP-46321 (0.1-1000 nM) for 72 hours. Cell viability was measured using a luminescent assay, and CC50 values were determined [1] |
| Animal Protocol |
Mice; ETP-46321 (50 mg/kg, p.o.) is administered to BALB/C mice every day for three weeks. Four mice from each treatment group had their tumor volumes measured and compared to the initial volume at the start of the treatment. MCF-7 xenograft model: Female BALB/c nude mice (4-6 weeks old) were subcutaneously implanted with 5×10⁶ MCF-7 cells. When tumors reached ~100 mm³, mice were randomly divided into vehicle control, ETP-46321 30 mg/kg, and 60 mg/kg groups (n=6 per group). The drug was dissolved in 0.5% methylcellulose + 0.2% Tween 80 and administered by oral gavage once daily for 21 days. Tumor volume was measured every 3 days using calipers, and tumor weight was recorded at euthanasia. Tumor tissues were collected for western blot analysis [1] - Ramos xenograft model: Female BALB/c nude mice (4-6 weeks old) were subcutaneously implanted with 5×10⁶ Ramos cells. When tumors reached ~100 mm³, mice were assigned to vehicle control, ETP-46321 20 mg/kg, and 40 mg/kg groups (n=6 per group). Drug formulation and administration were the same as the MCF-7 model, with treatment lasting 14 days. Tumor volume and weight were measured, and no additional tissue analysis was performed [1] |
| ADME/Pharmacokinetics |
Oral bioavailability: In rats, oral administration of ETP-46321 (10 mg/kg) resulted in an oral bioavailability of ~72% [1] - Plasma half-life (t1/2): In rats, t1/2 = 4.6 ± 0.5 hours (oral 10 mg/kg); in mice, t1/2 = 3.8 ± 0.4 hours (oral 30 mg/kg) [1] - Peak plasma concentration (Cmax): In rats, oral 10 mg/kg achieved Cmax = 1.8 ± 0.2 μg/mL at 1.2 ± 0.3 hours post-dosing; in mice, oral 30 mg/kg achieved Cmax = 3.2 ± 0.4 μg/mL at 1.0 ± 0.2 hours [1] - Area under the plasma concentration-time curve (AUC0-∞): In rats, AUC0-∞ = 9.5 ± 1.1 μg·h/mL (oral 10 mg/kg); in mice, AUC0-∞ = 15.8 ± 1.8 μg·h/mL (oral 30 mg/kg) [1] - Volume of distribution (Vd/F): In rats, Vd/F = 9.2 ± 1.2 L/kg (oral 10 mg/kg) [1] - Clearance (CL/F): In rats, CL/F = 18 ± 2 mL/min/kg (oral 10 mg/kg) [1] - Metabolism: ETP-46321 is metabolized primarily via oxidation in the liver, with minimal contribution from CYP450 isoforms (CYP3A4-mediated metabolism accounts for <10% of total metabolism) [1] - Excretion: In rats, ~65% of the oral dose was excreted in feces (mainly as metabolites) and ~25% in urine (parent drug and metabolites) within 72 hours [1] |
| Toxicity/Toxicokinetics |
In vitro cytotoxicity: ETP-46321 exhibited CC50 > 1 μM in normal human mammary epithelial cells (HMEC) and peripheral blood mononuclear cells (PBMCs) [1] - Acute toxicity in rats: Single oral administration of ETP-46321 up to 200 mg/kg did not cause mortality or overt toxicity (lethargy, weight loss, behavioral abnormalities) [1] - Chronic toxicity in mice: Repeated oral administration of ETP-46321 (60 mg/kg/day for 21 days) did not induce significant changes in hematological parameters (RBC, WBC, platelets) or serum biochemical markers (ALT, AST, creatinine, BUN) [1] - Plasma protein binding: ETP-46321 exhibited plasma protein binding of 94 ± 2% in rat plasma, 93 ± 3% in mouse plasma, and 92 ± 2% in human plasma (equilibrium dialysis) [1] |
| References |
[1]. Identification of ETP-46321, a potent and orally bioavailable PI3K α, δ inhibitor. Bioorg Med Chem Lett. 2012 May 15;22(10):3460-6. |
| Additional Infomation |
ETP-46321 is a potent, orally bioavailable small-molecule inhibitor of phosphatidylinositol 3-kinase α (PI3Kα) and δ (PI3Kδ) [1] - The therapeutic mechanism of ETP-46321 involves selective inhibition of PI3Kα and PI3Kδ isoforms, blocking the PI3K/Akt signaling pathway, which is aberrantly activated in many human cancers (e.g., breast cancer, B-cell lymphoma). This leads to G1 phase cell cycle arrest, inhibition of cancer cell proliferation, and induction of apoptosis [1] - ETP-46321 was developed as a potential therapeutic agent for the treatment of PI3Kα/δ-dependent solid tumors and hematological malignancies [1] - Preclinical data demonstrate significant in vitro antiproliferative activity against PI3Kα/δ-dependent cancer cell lines, potent in vivo antitumor efficacy in xenograft models, and favorable pharmacokinetic profiles (good oral bioavailability, moderate half-life, low clearance) [1] - The moderate selectivity of ETP-46321 for PI3Kα/δ over other PI3K isoforms minimizes off-target effects, contributing to its good tolerability in preclinical studies [1] |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.1117 mL | 10.5585 mL | 21.1171 mL | |
| 5 mM | 0.4223 mL | 2.1117 mL | 4.2234 mL | |
| 10 mM | 0.2112 mL | 1.0559 mL | 2.1117 mL |