Physicochemical Properties
| Molecular Formula | C18H23CLF2N4O2 |
| Molecular Weight | 400.8548 |
| Exact Mass | 400.147 |
| Elemental Analysis | C, 53.93; H, 5.78; Cl, 8.84; F, 9.48; N, 13.98; O, 7.98 |
| CAS # | 1950569-11-5 |
| Related CAS # | 1351438-26-0;1950569-11-5 (HCl); |
| PubChem CID | 121418768 |
| Appearance | White to off-white solid powder |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 6 |
| Rotatable Bond Count | 6 |
| Heavy Atom Count | 27 |
| Complexity | 463 |
| Defined Atom Stereocenter Count | 0 |
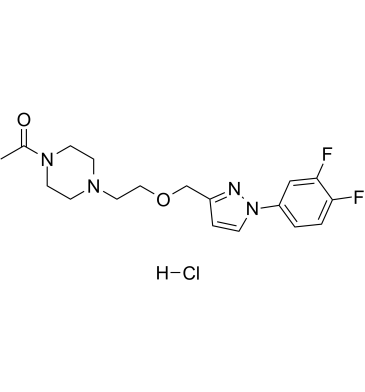
| SMILES | Cl.FC1=C(C=CC(=C1)N1C=CC(COCCN2CCN(C(C)=O)CC2)=N1)F |
| InChi Key | YJZGDOPAALDWAT-UHFFFAOYSA-N |
| InChi Code | InChI=1S/C18H22F2N4O2.ClH/c1-14(25)23-8-6-22(7-9-23)10-11-26-13-15-4-5-24(21-15)16-2-3-17(19)18(20)12-16;/h2-5,12H,6-11,13H2,1H3;1H |
| Chemical Name | 1-(4-(2-((1-(3,4-difluorophenyl)-1H-pyrazol-3-yl)methoxy)ethyl)piperazin-1-yl)ethan-1-one hydrochloride |
| Synonyms | EST64454 hydrochlorideEST-64454EST64454 EST 64454 |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month Note: Please store this product in a sealed and protected environment, avoid exposure to moisture. |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
σ1 receptor (Ki = 0.8 nM in radioligand binding assay); σ2 receptor (Ki > 1000 nM, > 1250-fold selectivity over σ2) [1] |
| ln Vitro |
σ1 receptor binding affinity: EST64454 HCl binds to human σ1 receptor with high affinity (Ki = 0.8 nM) and exhibits exceptional selectivity (> 1250-fold) over σ2 receptor (Ki > 1000 nM). It shows no significant binding to 45 other receptors/ion channels (e.g., μ/δ/κ opioid receptors, TRPV1, NaV1.7) at 10 μM [1] - σ1 receptor functional antagonism: In NG108-15 cells (expressing endogenous σ1 receptors), the compound antagonizes (+)-pentazocine (σ1 agonist)-induced Ca²⁺ mobilization with an IC50 of 2.3 μM. At 10 μM, it inhibits σ1 agonist-mediated Ca²⁺ response by 90% [1] - Inhibition of σ1-mediated signaling: EST64454 HCl (1–10 μM) blocks σ1 agonist-induced ERK1/2 phosphorylation in NG108-15 cells. At 5 μM, phosphorylated ERK1/2 levels are reduced by 75% compared to agonist-only control (western blot verification) [1] - High aqueous solubility: The compound exhibits excellent aqueous solubility (25 mg/mL in pH 7.4 buffer), significantly higher than other σ1 receptor antagonists (typically < 1 mg/mL) [1] |
| ln Vivo |
AUC0-∞, Vss, F%, and Cmax values of 771 ng/mL, 3.4 hours, 1431 ng·h/mL, and 69% were observed in male Wistar rats treated orally with EST64454 (10 mg/kg)[1]. After receiving 10 mg/kg of EST64454 intraperitoneally, male CD1 mice demonstrated the following: Cmax, t1/2, AUC0-∞, Vss, and F% of 1178 ng/mL, <1 hour, 2645 ng h/mL, 1.2 l/kg, and 60%, respectively [1]. Formalin-induced inflammatory pain inhibition: Male CD-1 mice were treated with EST64454 HCl (1–30 mg/kg, p.o.) 30 minutes before formalin (5%, 20 μL) hindpaw injection. The compound dose-dependently inhibits both early (0–5 min) and late (15–30 min) phases of formalin-induced licking/biting. At 30 mg/kg, early-phase response is reduced by 52% and late-phase by 68% [1] - Hot plate test analgesia: Female ICR mice were treated with EST64454 HCl (3–30 mg/kg, p.o.). The hot plate latency (55°C) was measured at 1, 2, 4 hours post-administration. At 10 mg/kg, the latency increased by 45% (2 hours post-dose) compared to vehicle, with analgesic effect lasting > 4 hours [1] - CCI-induced neuropathic pain relief: Male Sprague-Dawley rats with chronic constriction injury (CCI) of the sciatic nerve were treated with EST64454 HCl (3–30 mg/kg, p.o., qd) for 7 days. At 30 mg/kg, mechanical allodynia (von Frey filament) and thermal hyperalgesia (plantar test) were reduced by 62% and 58%, respectively, compared to vehicle-treated CCI rats [1] - No opioid-like side effects: Unlike morphine, EST64454 HCl (30 mg/kg, p.o.) did not induce tolerance (after 7-day repeated administration) or physical dependence (no withdrawal signs after naloxone challenge) in mice [1] |
| Enzyme Assay |
σ1 receptor radioligand binding assay: Recombinant human σ1 receptor was immobilized on microplates. Serial dilutions of EST64454 HCl (0.01 nM–10 μM) and [³H]-(+)-pentazocine (radiolabeled σ1 ligand) were co-incubated with the receptor at 25°C for 120 minutes. Unbound ligands were removed by washing, and bound radioactivity was measured with a scintillation counter. Ki value was calculated using competitive binding analysis [1] - σ2 receptor selectivity assay: Parallel assays were performed with recombinant human σ2 receptor and [³H]-DTG (radiolabeled σ2 ligand). EST64454 HCl at concentrations up to 10 μM showed < 0.1% displacement of [³H]-DTG, confirming σ1/σ2 selectivity [1] - Broad receptor/ion channel selectivity screening: The compound was tested for binding to a panel of 45 targets (opioid receptors, ion channels, GPCRs) using radioligand binding or functional assays. Binding/inhibition rate < 10% at 10 μM confirmed no off-target activity [1] |
| Cell Assay |
Ca²⁺ mobilization antagonism assay: NG108-15 cells were seeded in 96-well plates (3×10⁴ cells/well) and incubated overnight. Cells were loaded with a Ca²⁺-sensitive fluorescent dye for 60 minutes at 37°C. Serial dilutions of EST64454 HCl (0.1 μM–50 μM) were added, followed by (+)-pentazocine (10 μM, σ1 agonist). Fluorescence intensity (excitation/emission = 485/525 nm) was measured in real-time, and IC50 was derived from dose-response curves of Ca²⁺ response inhibition [1] - ERK1/2 phosphorylation assay: NG108-15 cells were seeded in 6-well plates (5×10⁵ cells/well) and pretreated with EST64454 HCl (1–10 μM) for 30 minutes, then stimulated with (+)-pentazocine (10 μM) for 15 minutes. Cells were lysed, proteins were separated by SDS-PAGE, transferred to membranes, and probed with anti-phospho-ERK1/2, anti-ERK1/2, and anti-β-actin (loading control) antibodies [1] |
| Animal Protocol |
Animal/Disease Models: Male Wistar rat (250-300 g) [1] Doses: 10 mg/kg Route of Administration: Po (pharmacokinetic/PK/PK analysis) Experimental Results: Cmax, t1/2, AUC0-∞, Vss and F% were 771 ng/mL, 3.4 hrs (hrs (hours)), 1431 ng h/mL, 4.4 l/kg and 69%. Formalin-induced pain model: Male CD-1 mice (6–8 weeks old, n=8 per group) were randomized to vehicle or EST64454 HCl groups (1, 3, 10, 30 mg/kg). The compound was dissolved in water (due to high solubility) and administered orally 30 minutes before hindpaw injection of 5% formalin (20 μL). The total time of hindpaw licking/biting was recorded during 0–5 min (early phase) and 15–30 min (late phase) [1] - Hot plate test: Female ICR mice (n=8 per group) were treated with EST64454 HCl (3, 10, 30 mg/kg, p.o.) or vehicle. The hot plate was maintained at 55°C, and paw withdrawal latency was measured before dosing and at 1, 2, 4 hours post-administration. A cutoff time of 30 seconds was used to avoid tissue damage [1] - CCI-induced neuropathic pain model: Male Sprague-Dawley rats (n=6 per group) underwent CCI surgery on the right sciatic nerve. Seven days post-surgery, rats were treated with EST64454 HCl (3, 10, 30 mg/kg, p.o.) once daily for 7 days. Mechanical allodynia was assessed using von Frey filaments (50% paw withdrawal threshold), and thermal hyperalgesia using a plantar test (paw withdrawal latency) [1] - Tolerance and dependence studies: Mice were treated with EST64454 HCl (30 mg/kg, p.o.) or morphine (10 mg/kg, s.c.) once daily for 7 days. Analgesic tolerance was evaluated by hot plate test on day 1 and day 7. Physical dependence was assessed by administering naloxone (5 mg/kg, i.p.) 24 hours after the last dose, and withdrawal signs (jumping, paw tremor) were counted for 30 minutes [1] - Pharmacokinetic study: Male Sprague-Dawley rats (n=3 per time point) were administered a single oral dose of EST64454 HCl (10 mg/kg, dissolved in water). Blood samples were collected at 0.25, 0.5, 1, 2, 4, 8, 12, 24 hours post-administration. Plasma drug concentrations were quantified by LC-MS/MS, and pharmacokinetic parameters were calculated using non-compartmental analysis [1] |
| ADME/Pharmacokinetics |
Oral bioavailability: In Sprague-Dawley rats, the oral bioavailability of EST64454 HCl is 85% following a single 10 mg/kg oral dose [1] - Plasma pharmacokinetics: Peak plasma concentration (Cmax) = 4.2 μM (Tmax = 1 hour), elimination half-life (t1/2) = 4.2 hours, AUC₀₋₂₄h = 22.8 μM·h [1] - Tissue distribution: Twenty-four hours after oral administration, the compound distributes widely to tissues relevant to pain signaling: brain (tissue/plasma ratio = 1.8), spinal cord (2.1), and dorsal root ganglia (2.5). Low accumulation in liver (1.2) and kidneys (1.1) [1] - Solubility and stability: Aqueous solubility = 25 mg/mL (pH 7.4 buffer), > 50-fold higher than clinical σ1 antagonists. Stable in simulated gastric fluid (pH 1.2) and intestinal fluid (pH 6.8) for 24 hours [1] - Metabolism: In vitro liver microsome assays show metabolism via CYP3A4-mediated oxidation, with 65% of parent compound remaining after 2 hours. No inhibition of CYP isoforms (CYP1A2, 2C9, 2C19, 2D6, 3A4) at concentrations up to 50 μM [1] - Excretion: In rats, 70% of the administered dose is excreted in feces (45% parent, 25% metabolites) and 25% in urine (15% parent, 10% metabolites) within 72 hours [1] |
| Toxicity/Toxicokinetics |
Acute toxicity: Single oral doses of EST64454 HCl up to 300 mg/kg in rats and mice show no mortality or acute toxicity signs (lethargy, ataxia, loss of appetite). LD50 > 300 mg/kg in both species [1] - Repeat-dose toxicity: Rats treated with EST64454 HCl (10, 50, 200 mg/kg, p.o., qd) for 28 days show no significant changes in body weight, hematological/biochemical parameters (ALT, AST, creatinine, BUN), or histopathology of major organs (brain, liver, kidney, heart, spinal cord) [1] - Plasma protein binding: In vitro assay shows EST64454 HCl binds to human plasma proteins at a rate of 92% [1] - CNS safety: No sedative or motor-impairing effects in mice at doses up to 30 mg/kg (rotarod test: no significant reduction in fall latency vs. vehicle) [1] |
| References |
[1]. EST64454: a Highly Soluble σ1 Receptor Antagonist Clinical Candidate for Pain Management. J Med Chem. 2020;63(23):14979-14988. |
| Additional Infomation |
Background: The σ1 receptor is a chaperone protein involved in pain signaling pathways, overactivated in inflammatory and neuropathic pain. σ1 antagonists offer a non-opioid approach to pain management, avoiding opioid-related tolerance, dependence, and overdose risks [1] - Mechanism of action: EST64454 HCl binds to the σ1 receptor’s allosteric site, blocking its interaction with downstream signaling partners (e.g., IP3 receptors, ERK). This inhibits σ1-mediated amplification of pain signals in the CNS and peripheral nervous system [1] - Therapeutic potential: As a clinical candidate for pain management, the compound effectively treats inflammatory (formalin) and neuropathic (CCI) pain with a favorable safety profile. Its high aqueous solubility enables oral administration and consistent bioavailability [1] - Advantages over existing agents: Compared to other σ1 antagonists, EST64454 HCl has superior solubility (> 25 mg/mL vs. < 1 mg/mL), higher oral bioavailability (85% vs. 40–60%), and longer analgesic duration (> 4 hours) [1] - Indication: Developed for the treatment of acute and chronic pain (e.g., inflammatory pain, neuropathic pain) as a non-opioid alternative [1] |
Solubility Data
| Solubility (In Vitro) |
H2O : ≥ 100 mg/mL (~249.47 mM) DMSO : ~100 mg/mL (~249.47 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.5 mg/mL (6.24 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.5 mg/mL (6.24 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.5 mg/mL (6.24 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 25.0 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 2.4947 mL | 12.4735 mL | 24.9470 mL | |
| 5 mM | 0.4989 mL | 2.4947 mL | 4.9894 mL | |
| 10 mM | 0.2495 mL | 1.2473 mL | 2.4947 mL |