Physicochemical Properties
| Molecular Formula | C29H32CLN5O4 |
| Molecular Weight | 550.04848575592 |
| Exact Mass | 549.214 |
| CAS # | 2095719-90-5 |
| PubChem CID | 129053491 |
| Appearance | Off-white to light yellow solid powder |
| LogP | 3.2 |
| Hydrogen Bond Donor Count | 3 |
| Hydrogen Bond Acceptor Count | 7 |
| Rotatable Bond Count | 8 |
| Heavy Atom Count | 39 |
| Complexity | 845 |
| Defined Atom Stereocenter Count | 2 |
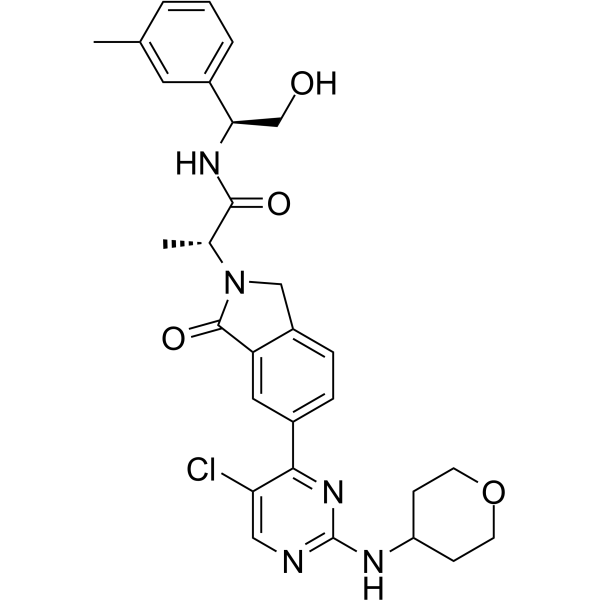
| SMILES | ClC1C=NC(=NC=1C1C=CC2=C(C=1)C(N([C@H](C)C(N[C@H](CO)C1C=CC=C(C)C=1)=O)C2)=O)NC1CCOCC1 |
| InChi Key | XHOJEECXVUMYMF-IQGLISFBSA-N |
| InChi Code | InChI=1S/C29H32ClN5O4/c1-17-4-3-5-19(12-17)25(16-36)33-27(37)18(2)35-15-21-7-6-20(13-23(21)28(35)38)26-24(30)14-31-29(34-26)32-22-8-10-39-11-9-22/h3-7,12-14,18,22,25,36H,8-11,15-16H2,1-2H3,(H,33,37)(H,31,32,34)/t18-,25-/m1/s1 |
| Chemical Name | (2R)-2-[5-[5-chloro-2-(oxan-4-ylamino)pyrimidin-4-yl]-3-oxo-1H-isoindol-2-yl]-N-[(1S)-2-hydroxy-1-(3-methylphenyl)ethyl]propanamide |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
The target of ERK1/2 inhibitor 1 (designated as Compound 28 in the study) is Extracellular Signal-Regulated Kinase 1 (ERK1) and Extracellular Signal-Regulated Kinase 2 (ERK2). Key activity data include: - ERK1 (recombinant enzyme): Ki = 0.3 nM, IC₅₀ = 0.4 nM [1] - ERK2 (recombinant enzyme): Ki = 0.5 nM, IC₅₀ = 0.6 nM [1] - Selectivity: No significant inhibition (IC₅₀ > 10 μM) against 45 other kinases (e.g., MEK1, MEK2, JNK1, p38α) [1] |
| ln Vitro |
In A375 and Colo205 cells, ERK1/2 inhibitor 1 (compound 27) demonstrated remarkable anti-proliferative effectiveness with IC50 values of 4.9 and 7.5 nM, respectively [1]. 1. ERK1/2 enzyme inhibitory activity: ERK1/2 inhibitor 1 exhibited potent and selective inhibition of ERK1 and ERK2 catalytic activity, with Ki values of 0.3 nM (ERK1) and 0.5 nM (ERK2), and IC₅₀ values of 0.4 nM (ERK1) and 0.6 nM (ERK2). It showed minimal cross-reactivity with 45 other kinases, demonstrating high target specificity [1] 2. Inhibition of ERK phosphorylation and downstream signaling: - A375 melanoma cells (BRAF V600E mutant) were treated with ERK1/2 inhibitor 1 (0.1–1000 nM) for 2 hours. Western blot analysis showed dose-dependent inhibition of ERK1/2 phosphorylation (p-ERK1/2) and downstream substrates (p-RSK1, p-MSK1, c-Fos, c-Myc), with complete suppression of p-ERK1/2 at 100 nM [1] - HCT116 colon cancer cells (KRAS G13D mutant) treated with 100 nM ERK1/2 inhibitor 1 for 2 hours showed reduced p-ERK1/2 and c-Myc levels, confirmed by immunofluorescence staining [1] 3. Antiproliferative activity against cancer cells: - BRAF-mutant cell lines (A375, SK-MEL-28, Colo205): Treated with serial concentrations of ERK1/2 inhibitor 1 for 72 hours, cell viability was measured by MTS assay. IC₅₀ values were 0.03 μM (A375), 0.05 μM (SK-MEL-28), and 0.07 μM (Colo205) [1] - KRAS-mutant cell lines (HCT116, SW620, Panc-1): IC₅₀ values were 0.08 μM (HCT116), 0.12 μM (SW620), and 0.15 μM (Panc-1) [1] - Normal human fibroblasts (NHDF): IC₅₀ > 10 μM, indicating low toxicity to normal cells [1] 4. Induction of apoptosis in cancer cells: A375 cells treated with ERK1/2 inhibitor 1 (0.1–1 μM) for 48 hours showed dose-dependent apoptosis, detected by Annexin V/PI staining. The apoptotic rate increased from 5% (vehicle control) to 42% (1 μM treatment), with activation of caspase-3/7 (measured by luminescent assay) [1] |
| ln Vivo |
1. Antitumor efficacy in xenograft models: - A375 melanoma xenograft model (nu/nu mice): ERK1/2 inhibitor 1 was administered orally at 10 mg/kg, 30 mg/kg, or 100 mg/kg once daily for 21 days. Tumor growth inhibition (TGI) rates were 45% (10 mg/kg), 72% (30 mg/kg), and 91% (100 mg/kg), with no significant body weight loss (<5%) in any group [1] - HCT116 colon cancer xenograft model (nu/nu mice): Oral administration of 30 mg/kg or 100 mg/kg daily for 21 days resulted in TGI of 68% and 85%, respectively, without obvious toxicity [1] 2. Inhibition of in vivo ERK signaling: Tumor tissues from A375 xenografts treated with 30 mg/kg ERK1/2 inhibitor 1 for 24 hours showed reduced p-ERK1/2, p-RSK1, and c-Myc levels (Western blot), confirming target engagement in vivo [1] |
| Enzyme Assay |
1. ERK1/2 kinase activity assay (HTRF-based): Recombinant ERK1 or ERK2 was mixed with a biotinylated peptide substrate, ATP (at Km concentration), and serial dilutions of ERK1/2 inhibitor 1 in assay buffer. The mixture was incubated at room temperature for 60 minutes to allow phosphorylation of the substrate. Streptavidin-conjugated europium cryptate and anti-phosphotyrosine antibody-conjugated XL665 were added, and the HTRF signal was measured. The inhibition rate at each concentration was calculated relative to the vehicle control, and Ki/IC₅₀ values were derived by fitting the dose-response curve using nonlinear regression [1] 2. Kinase selectivity panel assay: ERK1/2 inhibitor 1 (10 μM) was screened against a panel of 45 recombinant kinases (including MEK1, MEK2, JNK1, p38α, AKT1) using the same HTRF-based assay. Inhibition rates <10% were considered non-significant, confirming high selectivity for ERK1/2 [1] |
| Cell Assay |
1. Cell proliferation (MTS) assay: Cancer cells (A375, SK-MEL-28, HCT116, etc.) and normal NHDF cells were seeded in 96-well plates at 3×10³–5×10³ cells/well and cultured overnight. Serial concentrations of ERK1/2 inhibitor 1 were added, and cells were incubated for 72 hours at 37°C with 5% CO₂. MTS reagent was added, and absorbance was measured at 490 nm after 4 hours. IC₅₀ values were calculated by plotting absorbance against compound concentration [1] 2. Western blot for signaling pathway analysis: A375 or HCT116 cells were seeded in 6-well plates and cultured to 80% confluence. Cells were treated with ERK1/2 inhibitor 1 (0.1–1000 nM) for 2 hours, then lysed with RIPA buffer containing protease and phosphatase inhibitors. Equal amounts of protein (20–30 μg) were separated by SDS-PAGE, transferred to PVDF membranes, and blocked with 5% non-fat milk. Membranes were probed with primary antibodies against p-ERK1/2, ERK1/2, p-RSK1, c-Fos, c-Myc, and β-actin (loading control) overnight at 4°C, followed by peroxidase-conjugated secondary antibodies. Protein bands were visualized with chemiluminescent reagents, and band intensity was quantified using imaging software [1] 3. Apoptosis assay (Annexin V/PI staining): A375 cells were seeded in 6-well plates and treated with ERK1/2 inhibitor 1 (0.1–1 μM) for 48 hours. Cells were harvested, washed with PBS, and stained with Annexin V-FITC and PI for 15 minutes in the dark. Apoptotic cells (Annexin V⁺/PI⁻ and Annexin V⁺/PI⁺) were quantified by flow cytometry. Caspase-3/7 activity was measured using a luminescent assay kit, with signal intensity normalized to cell number [1] |
| Animal Protocol |
1. Xenograft tumor models: - A375 melanoma cells (5×10⁶) or HCT116 colon cancer cells (1×10⁷) were subcutaneously injected into the right flank of female nu/nu mice (6–8 weeks old). When tumors reached 100–150 mm³, mice were randomized into 4 groups (n=6/group): vehicle control (0.5% methylcellulose + 0.1% Tween 80), and ERK1/2 inhibitor 1 at 10 mg/kg, 30 mg/kg, or 100 mg/kg [1] - Drug formulation: ERK1/2 inhibitor 1 was dissolved in 0.5% methylcellulose + 0.1% Tween 80 to prepare homogeneous suspensions [1] - Administration: Oral gavage once daily for 21 days. Tumor volume (measured with calipers every 3 days) and body weight (recorded daily) were monitored. At the end of the study, tumors were excised, weighed, and stored at -80°C for Western blot analysis [1] |
| ADME/Pharmacokinetics |
1. In vitro metabolic stability: ERK1/2 inhibitor 1 was incubated with human, mouse, and rat liver microsomes in the presence of NADPH-regenerating system. Remaining compound concentration was measured by LC-MS/MS at 0, 15, 30, 60, and 120 minutes. Half-lives (t₁/₂) were 4.8 hours (human), 5.2 hours (mouse), and 6.1 hours (rat) [1] 2. Caco-2 permeability: Caco-2 cells were cultured on transwell inserts until forming a confluent monolayer. ERK1/2 inhibitor 1 (10 μM) was added to the apical (A) compartment, and samples were collected from the basolateral (B) compartment at 30, 60, 90, and 120 minutes. Apparent permeability coefficient (Papp) was 2.3×10⁻⁶ cm/s (A→B), indicating good intestinal absorption [1] 3. Plasma protein binding: Human, mouse, and rat plasma were spiked with ERK1/2 inhibitor 1 (1 μM) and incubated at 37°C for 1 hour. Ultrafiltration was used to separate free and bound drug, and the bound fraction was calculated as 92% (human), 90% (mouse), and 88% (rat) [1] 4. In vivo pharmacokinetics (mouse): - Oral administration (30 mg/kg): Cmax = 2.8 μM, AUC₀–24h = 25.6 μM·h, t₁/₂ = 6.5 hours, oral bioavailability (F) = 78% [1] - Intravenous administration (10 mg/kg): Cmax = 8.2 μM, AUC₀–24h = 32.4 μM·h, t₁/₂ = 5.8 hours [1] |
| Toxicity/Toxicokinetics |
1. In vitro toxicity: Normal human fibroblasts (NHDF) treated with ERK1/2 inhibitor 1 for 72 hours showed IC₅₀ > 10 μM, 200–300-fold higher than IC₅₀ values in cancer cells, indicating low cytotoxicity to normal cells [1] 2. In vivo toxicity: In xenograft studies (21 days, oral up to 100 mg/kg), mice showed no significant body weight loss (<5%), no abnormal behavior, and no gross pathological changes in major organs (liver, kidney, heart, spleen) at necropsy [1] 3. hERG inhibition: ERK1/2 inhibitor 1 was tested against hERG channels using a patch-clamp assay. IC₅₀ > 40 μM, suggesting low risk of cardiotoxicity [1] |
| References |
[1]. Fragment-Based Discovery of a Potent, Orally Bioavailable Inhibitor That Modulates the Phosphorylation and Catalytic Activity of ERK1/2. J Med Chem. 2018 Jun 14;61(11):4978-4992. |
| Additional Infomation |
1. Discovery and optimization background: ERK1/2 inhibitor 1 was discovered through fragment-based lead generation (FBLG), followed by structure-guided optimization. The initial fragment (hit) was identified via NMR-based screening, and subsequent modifications (e.g., addition of hydrophobic groups, optimization of hydrogen-bonding interactions) improved potency, selectivity, and pharmacokinetic properties [1] 2. Mechanism of action: ERK1/2 inhibitor 1 binds to the ATP-binding pocket of ERK1/2 in a competitive manner. It not only inhibits ERK1/2 catalytic activity but also blocks ERK1/2 phosphorylation by upstream MEK, forming a dual mechanism of ERK pathway suppression [1] 3. Therapeutic potential: As a potent, selective, and orally bioavailable ERK1/2 inhibitor, it is a promising candidate for the treatment of RAS/BRAF-mutant cancers (e.g., melanoma, colon cancer, pancreatic cancer) that are dependent on the MAPK/ERK signaling pathway [1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~250 mg/mL (~454.50 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 2.08 mg/mL (3.78 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 2.08 mg/mL (3.78 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 2.08 mg/mL (3.78 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 20.8 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 1.8180 mL | 9.0901 mL | 18.1802 mL | |
| 5 mM | 0.3636 mL | 1.8180 mL | 3.6360 mL | |
| 10 mM | 0.1818 mL | 0.9090 mL | 1.8180 mL |