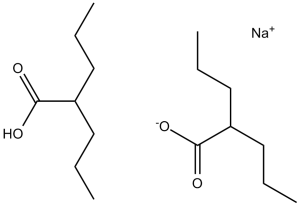
Divalproex Sodium, consisting of sodium valproate and valproic acid in a 1:1 molar ratio in an enteric coated form, is a potent HDAC inhibitor used in the treatment for epilepsy/seizures.
Physicochemical Properties
| Molecular Formula | C8H16O2.C8H15O2.NA | |
| Molecular Weight | 310.41 | |
| Exact Mass | 310.212 | |
| CAS # | 76584-70-8 | |
| Related CAS # | Valproic acid;99-66-1; Valproic acid sodium;1069-66-5;Valproic acid-d4;87745-17-3;Valproic acid-d6;87745-18-4;Valproic acid-d15;362049-65-8;Valproic acid (sodium)(2:1);76584-70-8;Valproic acid-d4 sodium;Valproic acid-d4-1;345909-03-7 | |
| PubChem CID | 23663956 | |
| Appearance | Typically exists as solid at room temperature | |
| Boiling Point | 220ºC at 760 mmHg | |
| Melting Point | 222ºC | |
| Flash Point | 116.6ºC | |
| LogP | 3.24 | |
| Hydrogen Bond Donor Count | 1 | |
| Hydrogen Bond Acceptor Count | 4 | |
| Rotatable Bond Count | 10 | |
| Heavy Atom Count | 21 | |
| Complexity | 192 | |
| Defined Atom Stereocenter Count | 0 | |
| SMILES | [Na+].[O-]C(C([H])(C([H])([H])C([H])([H])C([H])([H])[H])C([H])([H])C([H])([H])C([H])([H])[H])=O.O([H])C(C([H])(C([H])([H])C([H])([H])C([H])([H])[H])C([H])([H])C([H])([H])C([H])([H])[H])=O |
|
| InChi Key | MSRILKIQRXUYCT-UHFFFAOYSA-M | |
| InChi Code | InChI=1S/2C8H16O2.Na/c2*1-3-5-7(6-4-2)8(9)10;/h2*7H,3-6H2,1-2H3,(H,9,10);/q;;+1/p-1 | |
| Chemical Name | sodium;2-propylpentanoate;2-propylpentanoic acid | |
| Synonyms |
|
|
| HS Tariff Code | 2934.99.9001 | |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
|
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | HDAC1 ( IC50 = 400 μM ); HDAC1 ( IC50 = 0.5-2 mM ); HDAC2; Autophagy; Mitophagy |
| ln Vitro | In a dose- and time-dependent way, valproic acid (VPA) (0–15 mM; 24 and 72 h) suppresses the proliferation of Hela cells[1]. The activity of nuclear, cytosolic, and total HDACs is markedly reduced by valproic acid (10 mM; 24 h)[1]. The percentage of sub-G1 cells in HeLa cells rises when valproic acid (0–15 mM; 24 h) generates a G1/M phase arrest at 10 mM and a G1 phase arrest at 1-3 mM. Necrosis, apoptosis, and the release of lactate dehydrogenase (LDH) are additional effects of valproic acid[1]. Lithium works in concert with valproic acid (0–20 mM; 24 h) to stimulate Tcf/Lef-dependent transcription[2]. Neuro2A cells' β-catenin levels are elevated by valproic acid (0–5 mM; 0–18 h)[2]. Hepatocyte AMPK and ACC phosphorylation is stimulated by valproic acid (0–2 mM; 0–24 h)[5]. For two days, valproic acid (0 -10 mM) inhibits the generation of NE tumor markers in SCLC cells while inducing Notch1 signaling and morphologic differentiation[6]. |
| ln Vivo | In mice transplanted with Kasumi-1 cells, valproic acid (VPA) (500 mg/kg; ip; daily for 12 days) suppresses tumor angiogenesis[3]. Rats with valproic acid (350 mg/kg; ip; once) exhibit improved social behavior [4]. In obese mice, valproic acid (0.26% w/v; po via drinking water; 14 days) reduces blood glucose, hepatic fat formation, and liver mass without causing hepatotoxicity[5]. |
| Enzyme Assay | Valproic acid is widely used to treat epilepsy and bipolar disorder and is also a potent teratogen, but its mechanisms of action in any of these settings are unknown. We report that valproic acid activates Wntdependent gene expression, similar to lithium, the mainstay of therapy for bipolar disorder. Valproic acid, however, acts through a distinct pathway that involves direct inhibition of histone deacetylase (IC(50) for HDAC1 = 0.4 mm). At therapeutic levels, valproic acid mimics the histone deacetylase inhibitor trichostatin A, causing hyperacetylation of histones in cultured cells. Valproic acid, like trichostatin A, also activates transcription from diverse exogenous and endogenous promoters. Furthermore, valproic acid and trichostatin A have remarkably similar teratogenic effects in vertebrate embryos, while non-teratogenic analogues of valproic acid do not inhibit histone deacetylase and do not activate transcription. Based on these observations, we propose that inhibition of histone deacetylase provides a mechanism for valproic acid-induced birth defects and could also explain the efficacy of valproic acid in the treatment of bipolar disorder.[2] |
| Cell Assay |
Cell Viability Assay[1] Cell Types: HeLa cells Tested Concentrations: 0, 1, 3, 5, 10 and 15 mM Incubation Duration: 24 and 72 h Experimental Results: HeLa cell growth was dose- and time-dependently diminished with an IC50 of ~10 and 4 mM at 24 and 72 h. Western Blot Analysis[1][2][5] Cell Types: HeLa cells, Neuro2A cells or primary mouse hepatocytes Tested Concentrations: 10 mM (HeLa); 0, 2, and 5 mM (Neuro2A); 0.2, 0.4, 0.8, 1.2 and 2 mM (hepatocytes) Incubation Duration: 24 h (HeLa ); 0-18 h (Neuro2A); 0-24 h (hepatocytes) Experimental Results: Increased the form of acetylated histone 3. decreased PARP, induced cleavage PARP, and downregulated Bcl-2. Increased β-catenin levels. Increased the phosphorylation of AMPK and ACC. Cell Cycle Analysis[1] Cell Types: HeLa cells Tested Concentrations: 0, 1, 3, 5, 10 and 15 mM Incubation Duration: 24 h Experimental Results: Induced a G1 phase arrest at 1–3 mM, Dramatically induced a G2/M phase arrest at 10 mM, and increased the percentage of sub-G1 cells in HeLa cells in a dose-dependent manner at 24 h. |
| Animal Protocol |
Animal/Disease Models: Female BALB/c nude mice, Kasumi-1 tumor model[3] Doses: 500 mg/kg Route of Administration: intraperitoneal (ip)injection, daily for 12 days Experimental Results: Inhibited tumor growth and tumor angiogenesis. Inhibited the mRNA and protein expression of VEGF, VEGFR2 and bFGF. Inhibited HDAC activity and increased acetylation of histone H3. Enhanced the accumulation of hyperacetylated histone H3 on VEGF promoters. Animal/Disease Models: Timed-pregnant Long Evans rats[4] Doses: 350 mg/kg Route of Administration: intraperitoneal (ip)injection, once Experimental Results: Demonstrated more social investigation and play fighting than control animals. Animal/Disease Models: Obese phenotype of ob/ob mice[5] Doses: 0.26% (w/v) Route of Administration: Oral via drinking water, 14 days Experimental Results: Revealed a marked reduction in the accumulation of fats in the liver as compared with the untreated mice, Dramatically diminished liver mass to body mass, diminished serum triglyceride concentrations, and did not induce hepatotoxicity. |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion Absorption The intravenous and oral forms of valproic acid are expected to produce the same AUC, Cmax, and Cmin at steady-state. The oral delayed-release tablet formulation has a Tmax of 4 hours. Differences in absorption rate are expected from other formulations but are not considered to be clinically important in the context of chronic therapy beyond impacting frequency of dosing. Differences in absorption may create earlier Tmax or higher Cmax values on initiation of therapy and may be affected differently by meals. The extended release tablet formulation had Tmax increase from 4 hours to 8 hours when taken with food. In comparison, the sprinkle capsule formulation had Tmax increase from 3.3 hours to 4.8 hours. Bioavailability is reported to be approximately 90% with all oral formulations with enteric-coated forms possibly reaching 100%. Route of Elimination Most drug is eliminated through hepatic metabolism, about 30-50%. The other major contributing pathway is mitochondrial β-oxidation, about 40%. Other oxidative pathways make up an additional 15-20%. Less than 3% is excreted unchanged in the urine. Volume of Distribution 11 L/1.73m2. Clearance 0.56 L/hr/m2 Pediatric patients between 3 months and 10 years of age have 50% higher clearances by weight. Pediatric patients 10 years of age or older approximate adult values. Valproic acid and its salt, sodium valproate, are excreted into human milk in low concentrations. Milk concentrations up to 15% of the corresponding level in the mother's serum have been measured. In two infants, serum levels of valporate were 1.5% and 6.0% of maternal values. Placenta transfer study in non-human primate (NHP) is one of the crucial components in the assessment of developmental toxicity because of the similarity between NHP and humans. To establish the method to determine placenta transfer in non-human primate, toxicokinetics of valproic acid (VPA), a drug used to treat epilepsy in pregnant women, were determined in pregnant cynomolgus monkeys. After mating, pregnancy-proven females were daily administered with VPA at dose levels of 0, 20, 60 and 180 mg/kg by oral route during the organogenesis period from gestation day (GD) 20 to 50. Concentrations of VPA and its metabolite, 4-ene-VPA, in maternal plasma on GDs 20 and 50, and concentrations of VPA and 4-ene-VPA in placenta, amniotic fluid and fetus on GD 50 were analyzed using LC/MS/MS. Following single oral administration of VPA to pregnant monkeys, concentrations of VPA and 4-ene-VPA were generally quantifiable in the plasma from all treatment groups up to 4-24 hours post-dose, demonstrating that VPA was absorbed and the monkeys were systemically exposed to VPA and 4-ene-VPA. After repeated administration of VPA to the monkeys, VPA was detected in amniotic fluid, placenta and fetus from all treatment groups, demonstrating that VPA was transferred via placenta and the fetus was exposed to VPA, and the exposures were increased with increasing dose. Concentrations of 4-ene-VPA in amniotic fluid and fetus were below the limit of quantification, but small amount of 4-ene-VPA was detected in placenta. In conclusion, pregnant monkeys were exposed to VPA and 4-ene-VPA after oral administration of VPA at dose levels of 20, 60 and 180 mg/kg during the organogenesis period. VPA was transferred via placenta and the fetus was exposed to VPA with dose-dependent exposure. The metabolite, 4-ene VPA, was not detected in both amniotic fluid and fetus, but small amount of 4-ene-VPA was detected in placenta. These results demonstrated that proper procedures to investigate placenta transfer in NHP, such as mating and diagnosis of pregnancy via examining gestational sac with ultrasonography, collection of amniotic fluid, placenta and fetus after Caesarean section followed by adequate bioanalysis and toxicokinetic analysis, were established in this study using cynomolugus monkeys. PMID:24278535 View More
Metabolism / Metabolites Most drug is metabolized to glucuronide conjugates (30-50%) of the parent drug or of metabolites. Another large portion is metabolized through mitochondrial β-oxidation (40%). The remainder of metabolism (15-20%) occurs through oxidation, hydroxylation, and dehydrogenation at the ω, ω1, and ω2 positions resulting in the formation of hydroxyls, ketones, carboxyls, a lactone metabolite, double bonds, and combinations. The aim of this study was to investigate the relationship between hepatotoxicity, levels of glucuronide conjugates of valproic acid (VPA), and the toxic metabolites of VPA (4-ene VPA and 2,4-diene VPA). /The study/ also examined whether hepatotoxicity could be predicted by the urinary excretion levels of VPA and its toxic metabolites. VPA was administrated orally in rats in amounts ranging from 20 mg/kg to 500 mg/kg. Free and total (free plus glucuronide conjugated) VPA, 4-ene VPA, and 2,4-diene VPA were quantified in urine and liver using gas chromatography-mass spectrometry. Serum levels of aspartate aminotransferase, alanine aminotransferase, and alpha-glutathione S-transferase (alpha-GST) were also determined to measure the level of hepatotoxicity. The serum alpha-GST level increased slightly at the 20 mg/kg dose, and substantially increased at the 100 and 500 mg/kg dose; aspartate aminotransferase and alanine aminotransferase levels did not change with the administration of increasing doses of VPA. The liver concentration of free 4-ene VPA and the urinary excretion of total 4-ene VPA were the only measures that correlated with the increase in the serum alpha-GST level (p < 0.094 and p < 0.023 respectively). From these results, /it is concluded/ that hepatotoxicity of VPA correlates with liver concentration of 4-ene VPA and can be predicted by the urinary excretion of total 4-ene VPA. PMID:19641884 BACKGROUND AND OBJECTIVE: Sodium valproate is a widely prescribed broad-spectrum antiepileptic drug. It shows high inter-individual variability in pharmacokinetics and pharmacodynamics and has a narrow therapeutic range. /This study/ evaluated the effects of polymorphic uridine diphosphate glucuronosyltransferase (UGT)1A6 (541A>G, 552A>C) metabolizing enzyme on the pharmacokinetics of sodium valproate in the patients with epilepsy who showed toxicity to therapy. METHODS: Genotype analysis of the patients was made with polymerase chain-restriction fragment length polymorphism (RFLP) with sequencing. Plasma drug concentrations were measured with reversed phase high-performance liquid chromatography (HPLC) and concentration-time data were analyzed by using a non-compartmental approach. RESULTS: The results of this study suggested a significant genotypic as well as allelic association with valproic acid toxicity for UGT1A6 (541A>G) or UGT1A6 (552A>C) polymorphic enzymes. The elimination half-life (t 1/2 = 40.2 hr) of valproic acid was longer and the clearance rate (CL = 917 mL/hr) was lower in the poor metabolizers group of UGT1A6 (552A>C) polymorphism who showed toxicity than in the intermediate metabolizers group (t 1/2= 35.5 hr, CL = 1,022 mL/hr) or the extensive metabolizers group (t 1/2= 25.4 hr, CL = 1,404 mL/hr). CONCLUSION: /These/ findings suggest that the UGT1A6 (552A>C) genetic polymorphism plays a significant role in the steady state concentration of valproic acid, and it thereby has an impact on the toxicity of the valproic acid used in the patients with epilepsy. PMID:23749495 Biotransformation /of valproic acid/ is primarily hepatic. Some metabolites may have pharmacologic or toxic activity. Rate of metabolism is faster in children and in patients concurrently using enzyme-inducing medications, such as phenytoin, phenobarbital, primidone, and carbamazepine. Valproate is metabolized almost entirely by the liver. In adult patients on monotherapy, 30- 50% of an administered dose appears in urine as a glucuronide conjugate. Mitochondrial -oxidation is the other major metabolic pathway, typically accounting for over 40% of the dose. Usually, less than 15-20% of the dose is eliminated by other oxidative mechanisms. Less than 3% of an administered dose is excreted unchanged in urine. Valproic acid has known human metabolites that include (2S,3S,4S,5R)-3,4,5-Trihydroxy-6-(2-propylpentanoyloxy)oxane-2-carboxylic acid, 4-Hydroxyvalproate, 3-Hydroxyvalproate, 5-Hydroxyvalproate, and 4-ene-valproate. Valproic acid is rapidly absorbed from gastrointestinal tract. Valproic acid is metabolized almost entirely by the liver. In adult patients on monotherapy, 30-50% of an administered dose appears in urine as a glucuronide conjugate. Mitochondrial oxidation is the other major metabolic pathway, typically accounting for over 40% of the dose. These products include 2-n-propylpent-2-enoic acid (delta 2,3 VPE) and several coenzyme A (CoA) derivatives including VPA-CoA, and delta 2,3 VPE-CoA. Usually, less than 15-20% of the dose is eliminated by other oxidative mechanisms. Less than 3% of an administered dose is excreted unchanged in urine (A308). Half Life: 9-16 hours (following oral administration of 250 mg to 1000 mg). Biological Half-Life 13-19 hours. The half-life in neonates ranges from 10-67 hours while the half-life in pediatric patients under 2 months of age ranges from 7-13 hours. In children the half-life of valproic acid alone is 10 to 11 hours; when other medications are added, half-life may be reduced to 8 to 9 hours. Half-lives of up to 30 hours have been reported in overdosage. International Programme on Chemical Safety (IPCS); Poisons Information Monograph: Valproic Acid (PIM 551) (1997) Available from, as of May 30, 2007: https://www.inchem.org/pages/pims.html Variable, from 6 to 16 hours; may be considerably longer in patients with hepatic function impairment, in the elderly, and in children up to 18 months of age; may be considerably shorter in patients receiving hepatic enzyme-inducing anticonvulsants. In one study, the half life in children under 10 days ranged from 10 to 67 hours compared to a range of 7 to 13 hours in children greater than 2 months. |
| Toxicity/Toxicokinetics |
Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Very little information is available on the clinical use of divalproex during breastfeeding. However, divalproex is rapidly metabolized in the body to the active drug valproic acid. Valproic acid levels in breastmilk are low and infant serum levels range from undetectable to low. Breastfeeding during valproic acid monotherapy does not appear to adversely affect infant growth or development, and breastfed infants had higher IQs and enhanced verbal abilities than nonbreastfed infants at 6 years of age in one study. A safety scoring system finds valproic acid possible to use during breastfeeding. If valproic acid is required by the mother, it is not necessarily a reason to discontinue breastfeeding. No definite adverse reactions to valproic acid in breastfed infants have been reported. Theoretically, breastfed infants are at risk for valproic acid-induced hepatotoxicity, so infants should be monitored for jaundice and other signs of liver damage during maternal therapy. A questionable case of thrombocytopenia has been reported, so monitor the infant for unusual bruising or bleeding. A rare case of infant baldness might have been caused by valproate in milk. Observe the infant for jaundice and unusual bruising or bleeding. Combination therapy with sedating anticonvulsants or psychotropics may result in infant sedation or withdrawal reactions. ◉ Effects in Breastfed Infants A mother with epilepsy was taking valproic acid 2.4 grams daily and primidone 250 mg 3 times daily during pregnancy and postpartum. During the second week postpartum, her breastfed infant was sedated. Breastfeeding was stopped and the drowsiness cleared. The sedation was possibly caused by primidone in breastmilk although valproic acid might have contributed by increasing primidone levels. Petechiae, thrombocytopenia, anemia, and mild hematuria occurred in a 2.5-month-old breastfed infant whose mother was taking valproic acid 600 mg twice daily. Blood hemoglobin and reticulocytes normalized between 12 and 19 days after discontinuing breastfeeding. The petechiae resolved 35 days after discontinuing breastfeeding and the infant's platelet count had almost reached the normal range by this time. By day 83, the infant's platelet count was well within the normal range. The authors believed the adverse effect to be caused by valproic acid in breastmilk. However, other authors believe that these symptoms were more likely caused by idiopathic thrombocytopenic purpura following a viral infection. Two breastfed infants aged 1 and 3 months whose mothers were taking valproic acid monotherapy 750 and 500 mg daily developed normally and had no abnormal laboratory values. Their plasma levels were 6% and 1.5% or their mother's serum levels, respectively. Six breastfed infants whose mothers were taking valproic acid 750 or 1000 mg daily had no adverse reactions to valproic acid in breastmilk. An exclusively breastfed infants whose mother was taking valproate 1.8 g, topiramate 300 mg, and levetiracetam 2 g, daily during pregnancy and lactation appeared healthy to the investigators throughout the 6- to 8-week study period. In a long-term study on infants exposed to anticonvulsants during breastfeeding, no difference in average intelligence quotient at 3 years of age was found between infants who were breastfed (n = 11) a median of 6 months and those not breastfed (n = 24) when their mothers were taking valproate monotherapy. At 6 years of age, extensive psychological and intelligence testing found that the breastfed infants had higher IQ values than the nonbreastfed infants. A prospective cohort study in Norway followed infants of mothers who took antiepileptic drugs during pregnancy and lactation and compared them to infants of mothers with untreated epilepsy and infants with fathers who took antiepileptics as control groups. Of the 223 mothers studied, 27 were taking valproate monotherapy. Infants were assessed at 6, 18 and 36 months of age. Continuous breastfeeding in children of women using antiepileptic drugs was associated with no greater impaired development than those with no breastfeeding or breastfeeding for less than 6 months. A woman with bipolar disorder who delivered twins and was taking sodium valproate in a therapeutic dosage was started on quetiapine 200 mg and olanzapine 15 mg at 11 pm daily after 20 days postpartum. She withheld breastfeeding during the night and discarded milk pumped at 7 am. She then breastfed her infants until 11 pm. The mother continued feeding the infants on this schedule for 15 months. Monthly follow-up of the infants indicated normal growth and neither the pediatricians nor the parents noted any adverse effects in the infants. The 4-month-old breastfed infant of a mother taking divalproex for bipolar disorder developed patchy hair loss. The extent of nursing and dosage of divalproex were not stated. Divalproex was discontinued and 2 months later, the infant’s hair was normal. The hair loss was possibly caused by valproate. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. |
| References |
[1]. Valproic acid inhibits the growth of HeLa cervical cancer cells via caspase-dependent apoptosis. Oncol Rep. 2013 Dec;30(6):2999-3005. [2]. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001 Sep 28;276(39):36734-41. [3]. Valproic acid inhibits tumor angiogenesis in mice transplanted with Kasumi 1 leukemia cells. Mol Med Rep. 2013 Nov 28. [4]. Acute prenatal exposure to a moderate dose of valproic acid increases social behavior and alters gene expression in rats. Int J Dev Neurosci. 2013 Dec;31(8):740-50. [5]. Valproic Acid Is a Novel Activator of AMP-Activated Protein Kinase and Decreases Liver Mass, Hepatic Fat Accumulation, and Serum Glucose in Obese Mice. Mol Pharmacol. 2014 Jan;85(1):1-10. [6]. Valproic acid induces Notch1 signaling in small cell lung cancer cells. J Surg Res. 2008 Jul;148(1):31-7. [7]. Valproic acid in association with highly active antiretroviral therapy for reducing systemic HIV-1 reservoirs: results from a multicentre randomized clinical study. HIV Med. 2012 May;13(5):291-6. |
| Additional Infomation |
Valproate semisodium is a mixture of valproic acid and its sodium salt in a 1:1 molar ratio. It is used for the management and treatment of seizure disorders, mania, and prophylactic treatment of migraine headache. It has a role as an antimanic drug, an anticonvulsant and a GABA agent. It contains a valproic acid and a sodium valproate. Divalproex Sodium is a stable coordination compound comprised of sodium valproate and valproic acid with anticonvulsant and antiepileptic activities. Divalproex dissociates to the valproate ion in the gastrointestinal tract. This agent binds to and inhibits gamma-aminobutyric acid (GABA) transaminase and its anticonvulsant activity may be exerted by increasing brain concentration of GABA and by inhibiting enzymes that catabolize GABA or block the reuptake of GABA into glia and nerve endings. Divalproex may also work by suppressing repetitive neuronal firing through inhibition of voltage-sensitive sodium channels. A fatty acid with anticonvulsant and anti-manic properties that is used in the treatment of EPILEPSY and BIPOLAR DISORDER. The mechanisms of its therapeutic actions are not well understood. It may act by increasing GAMMA-AMINOBUTYRIC ACID levels in the brain or by altering the properties of VOLTAGE-GATED SODIUM CHANNELS. See also: Valproic Acid (has active moiety). |
Solubility Data
| Solubility (In Vitro) |
|
|||
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2215 mL | 16.1077 mL | 32.2155 mL | |
| 5 mM | 0.6443 mL | 3.2215 mL | 6.4431 mL | |
| 10 mM | 0.3222 mL | 1.6108 mL | 3.2215 mL |