Desogestrel (Org-2969, Desogen; Org2969, Desogestrelum, Org 2969, Cerazette) is an approved synthetic progestin derivative used in female birth control pills as an oral contraceptive. Additionally, it has been applied to women's menopausal symptoms. When combined with an estrogen, desogestrel can be used as the progestogenic ingredient in oral contraceptive agents.
Physicochemical Properties
| Molecular Formula | C22H30O |
| Molecular Weight | 310.47 |
| Exact Mass | 310.229 |
| Elemental Analysis | C, 85.11; H, 9.74; O, 5.15 |
| CAS # | 54024-22-5 |
| Related CAS # | 54024-22-5 |
| PubChem CID | 40973 |
| Appearance | White to off-white solid powder |
| Density | 1.1±0.1 g/cm3 |
| Boiling Point | 428.3±45.0 °C at 760 mmHg |
| Melting Point | 109-110ºC |
| Flash Point | 187.9±21.7 °C |
| Vapour Pressure | 0.0±2.3 mmHg at 25°C |
| Index of Refraction | 1.566 |
| LogP | 6.59 |
| Hydrogen Bond Donor Count | 1 |
| Hydrogen Bond Acceptor Count | 1 |
| Rotatable Bond Count | 2 |
| Heavy Atom Count | 23 |
| Complexity | 605 |
| Defined Atom Stereocenter Count | 6 |
| SMILES | O([H])[C@@]1(C#C[H])C([H])([H])C([H])([H])[C@@]2([H])[C@]3([H])C([H])([H])C([H])([H])C4=C([H])C([H])([H])C([H])([H])C([H])([H])[C@]4([H])[C@@]3([H])C(=C([H])[H])C([H])([H])[C@@]21C([H])([H])C([H])([H])[H] |
| InChi Key | RPLCPCMSCLEKRS-BPIQYHPVSA-N |
| InChi Code | InChI=1S/C22H30O/c1-4-21-14-15(3)20-17-9-7-6-8-16(17)10-11-18(20)19(21)12-13-22(21,23)5-2/h2,8,17-20,23H,3-4,6-7,9-14H2,1H3/t17-,18-,19-,20+,21-,22-/m0/s1 |
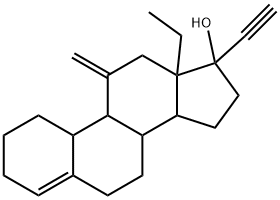
| Chemical Name | (8S,9S,10R,13S,14S,17R)-13-ethyl-17-ethynyl-11-methylidene-1,2,3,6,7,8,9,10,12,14,15,16-dodecahydrocyclopenta[a]phenanthren-17-ol |
| Synonyms | Cerazette; DESOGESTREL; Desogen; 54024-22-5; Cerazette; Desogestrelum; Org2969; ORG 2969; 13-Ethyl-11-methylene-18,19-dinor-17alpha-pregn-4-en-20-yn-17-ol; CHEBI:4453; Org-2969; Desogestrelum |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets |
- Progesterone Receptor (PR): As a progestin, Desogestrel exerts biological effects by binding to PR; specific Ki/EC50 values were not available in the abstract [2][3][4][5] - PHOX2B (Paired-Like Homeobox 2B): Desogestrel downregulates the expression of PHOX2B and its target genes (e.g., DBH, TH) in progesterone-responsive neuroblastoma cells; specific binding affinity (Ki) or regulatory activity (EC50) values were not available in the abstract [6] |
| ln Vitro |
- Metabolism experiments: In vitro metabolism of Desogestrel was investigated using liver microsomes or hepatocytes from several species (e.g., rat, dog, human). The main metabolite identified was 3-keto-desogestrel (3-KD), and the metabolic pathways involved cytochrome P450 (CYP) enzymes (specific CYP isoforms not detailed in the abstract). The metabolic rate and metabolite profile varied among species [1] - Neuroblastoma cell experiments: Desogestrel was treated to progesterone-responsive neuroblastoma cell lines (specific cell line not named in the abstract). After treatment, the mRNA and protein levels of PHOX2B (a transcription factor associated with neuroblastoma) and its downstream target genes (e.g., dopamine β-hydroxylase, DBH; tyrosine hydroxylase, TH) were significantly downregulated, as detected by quantitative real-time PCR (qPCR) and Western blot. This downregulation was dependent on progesterone receptor activation (confirmed by co-treatment with PR antagonists, which reversed the effect) [6] |
| ln Vivo |
Desogestrel is extensively metabolized in rats and dogs following oral administration; in rats, desogestrel is primarily metabolized at the C3, C5, C11, and C15 positions. Desogestrel's 15α-position is modified by adding a hydroxy group, which is then conjugated with glucuronic acid. Dogs metabolize desogestrel primarily at the C3 and C17 positions[1].
- Hormonal suppression in contraception: Two contraceptive regimens containing ethinyl estradiol (EE) plus Desogestrel (specific doses: e.g., EE 20μg + Desogestrel 150μg, EE 30μg + Desogestrel 150μg) were administered to healthy women (sample size ~50-100 per group) for 21 consecutive days. The regimens significantly suppressed luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels (suppression rate >70% vs. baseline) and inhibited ovulation (ovulation rate <5% in both groups). No significant difference in hormonal suppression efficacy was observed between the two dose regimens [2] - Lipid metabolism effects: Two progestogen-only pills (POPs) – one containing Desogestrel (75μg/day) and the other levonorgestrel (30μg/day) – were administered to healthy women (sample size ~30 per group) for 6 months. Compared with the levonorgestrel group, the Desogestrel group showed no significant change in total cholesterol, low-density lipoprotein (LDL)-cholesterol, or high-density lipoprotein (HDL)-cholesterol levels; however, a slight but non-significant increase in triglyceride levels was observed (mean increase ~0.1 mmol/L vs. baseline) [4] - Thyroid function effects: A progestin (including Desogestrel, specific dose not detailed in the abstract) was administered to female Wistar rats (n=10-15 per group) for 28 days. The treatment resulted in a slight increase in serum thyroid-stimulating hormone (TSH) levels (mean increase ~0.2 mIU/L vs. control) and no significant changes in free triiodothyronine (FT3) or free thyroxine (FT4) levels. The effect was reversible after drug withdrawal [7] - In vivo metabolism across species: Desogestrel was administered to several animal species (rat, dog, monkey; specific routes: oral, intravenous) and humans. After oral administration, Desogestrel was rapidly absorbed and metabolized to 3-KD (the major active metabolite). The plasma half-life (t1/2) of 3-KD was ~12-20 hours in humans, ~8-12 hours in rats, and ~15-25 hours in dogs. 3-KD was mainly distributed in reproductive tissues (e.g., uterus, ovaries) and liver, and excreted primarily via feces (60-70%) and urine (20-30%) [1][5] |
| Enzyme Assay |
- Liver microsome metabolism assay: Liver microsomes were prepared from different species (rat, dog, human). Desogestrel (final concentration 1-10 μM) was incubated with microsomes and NADPH (cofactor for CYP enzymes) at 37°C for 0-60 minutes. The reaction was terminated by adding acetonitrile. Metabolites (e.g., 3-KD) were separated and quantified using high-performance liquid chromatography (HPLC) or liquid chromatography-mass spectrometry (LC-MS). The metabolic rate was calculated based on the decrease in Desogestrel concentration over time, and the intrinsic clearance (CLint) was determined to compare metabolic activity across species [1] - PHOX2B target gene expression assay (qPCR): Total RNA was extracted from neuroblastoma cells treated with Desogestrel (0.1-10 μM) for 24-48 hours. RNA was reverse-transcribed into cDNA using reverse transcriptase. qPCR was performed using specific primers for PHOX2B, DBH, and TH (housekeeping gene: GAPDH as internal control). The reaction conditions included initial denaturation at 95°C for 5 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 30 seconds. The relative expression levels of target genes were calculated using the 2^(-ΔΔCt) method [6] - PHOX2B protein expression assay (Western blot): Total protein was extracted from Desogestrel-treated neuroblastoma cells. Protein concentration was determined using a BCA assay. Equal amounts of protein (20-50 μg) were separated by SDS-PAGE and transferred to PVDF membranes. Membranes were blocked with 5% non-fat milk for 1 hour, then incubated with primary antibodies against PHOX2B (1:1000 dilution) and GAPDH (1:5000 dilution) overnight at 4°C. After washing, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:2000 dilution) for 1 hour at room temperature. Protein bands were visualized using an ECL detection system, and band intensity was quantified using ImageJ software [6] |
| Cell Assay |
- Neuroblastoma cell culture and treatment: Progesterone-responsive neuroblastoma cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in a 5% CO2 incubator. Cells were seeded into 6-well plates (1×10^5 cells/well) and allowed to attach overnight. Desogestrel was dissolved in dimethyl sulfoxide (DMSO) and added to the medium at final concentrations of 0.1 μM, 1 μM, and 10 μM (DMSO concentration <0.1% to avoid cytotoxicity). For PR antagonist experiments, cells were pre-treated with a PR antagonist (1 μM) for 1 hour before adding Desogestrel. Cells were incubated for 24-48 hours, then harvested for RNA or protein extraction [6] - Cell viability assay (for neuroblastoma cells): After Desogestrel treatment (0.1-10 μM, 48 hours), cell viability was assessed using the MTT assay. MTT solution (5 mg/mL) was added to each well (final concentration 0.5 mg/mL) and incubated for 4 hours at 37°C. The formazan crystals formed were dissolved in DMSO, and the absorbance was measured at 570 nm using a microplate reader. No significant cytotoxicity was observed at the tested concentrations of Desogestrel (viability >90% vs. control) [6] |
| Animal Protocol |
Female Wistar rats, Female beagle dogs 56 μg/kg, 106 mg/kg (Rats); 67 μg/kg, 9.6 mg/kg(Dogs) oral administration - In vivo metabolism study in rats: Male Sprague-Dawley rats (n=6 per group, 250-300 g) were used. Desogestrel was administered via two routes: (1) oral gavage (dose: 10 mg/kg, dissolved in 0.5% carboxymethyl cellulose, CMC); (2) intravenous injection (dose: 2 mg/kg, dissolved in saline containing 5% DMSO). Blood samples (0.5 mL) were collected from the tail vein at 0, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours post-administration. Plasma was separated by centrifugation (3000 rpm, 10 minutes) and stored at -80°C. Desogestrel and its metabolite 3-KD in plasma were quantified by LC-MS. Tissues (liver, kidney, uterus, ovaries) were collected at 2 hours post-oral administration for metabolite distribution analysis [1] - Thyroid function study in Wistar rats: Female Wistar rats (n=12 per group, 180-220 g) were randomly divided into control and Desogestrel groups. The Desogestrel group received a daily oral dose of Desogestrel (0.1 mg/kg, dissolved in corn oil) via gavage for 28 days; the control group received corn oil alone. At the end of the treatment, rats were anesthetized with isoflurane, and blood samples were collected from the abdominal aorta. Serum was separated and analyzed for TSH, FT3, and FT4 levels using enzyme-linked immunosorbent assay (ELISA). Thyroid glands were harvested, fixed in 4% paraformaldehyde, and embedded in paraffin for histological examination (no significant pathological changes were observed) [7] - Contraceptive efficacy study in non-human primates (monkeys): Female rhesus monkeys (n=8 per group, 5-7 years old) were administered an oral contraceptive containing Desogestrel (150 μg/day) and EE (30 μg/day) for 21 days, followed by a 7-day drug-free period, for 3 consecutive cycles. Blood samples were collected twice weekly to measure LH, FSH, and progesterone levels (progesterone <1 ng/mL indicated ovulation inhibition). Ovarian ultrasound was performed once weekly to monitor follicle development (follicle diameter <10 mm indicated no ovulation). The contraceptive regimen inhibited ovulation in all monkeys during the treatment period [5] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion After oral administration, desogestrel is rapidly absorbed and it reaches a peak concentration of 2 ng/ml after 1.5 hours. The bioavailability of desogestrel is reported to be in the range of 60-80% and the reported AUC is of 3000 ng.h/ml. Almost all the administered dose is modified to the active metabolite, [etonogestrel]. The elimination of desogestrel is found to be mainly renal corresponding to about 6 times the dose eliminated in the bile. The elimination of desogestrel is only done as the metabolites and not as the unchanged drug and about 85% of the administered dose can be excreted as metabolites after 6-8 days. The apparent volume of distribution of desogestrel is of 1.5 L/kg. The metabolic clearance rate of desogestrel is reported to be of about 2 ml/min/kg. After oral dosing of Cerazette desogestrel (DSG) is rapidly absorbed and converted into etonogestrel (ENG). Under steady-state conditions, peak serum levels are reached 1.8 hours after tablet-intake and the absolute bioavailability of ENG is approximately 70%. In the third cycle of use after a single desogestrel and ethinyl estradiol tablet, maximum concentrations of 3-keto-desogestrel of 2,805 +/- 1,203 pg/mL (mean+/-SD) are reached at 1.4+/-0.8 hours. The area under the curve (AUC) is 33,858+/-11,043 pg/mL (hr) after a single dose. At steady state, attained from at least day 19 onwards, maximum concentrations of 5,840 +/-1,667 pg/mL are reached at 1.4+/-0.9 hours. The minimum plasma levels of 3-keto-desogestrel at steady state are 1,400+/-560 pg/mL. The AUC0-24 at steady state is 52,299+/-17,878 pg/mL (hr). The mean AUC0 for 3-keto-desogestrel at single dose is significantly lower than the mean AUC0-24 at steady state. This indicates that the kinetics of 3-keto-desogestrel are non-linear due to an increase in binding of 3-keto-desogestrel to sex hormone-binding globulin in the cycle, attributed to increased sex hormone-binding globulin levels which are induced by the daily administration of ethinyl estradiol. Sex hormone-binding globulin levels increased significantly in the third treatment cycle from day 1 (150+/-64 nmol/L) to day 21 (230+/-59 nmol/L). Etonogestrel is 95.5-99% bound to serum proteins, predominantly to albumin and to a lesser extent to sex hormone-binding globulin (SHBG). For more Absorption, Distribution and Excretion (Complete) data for DESOGESTREL (8 total), please visit the HSDB record page. Metabolism / Metabolites Desogestrel is rapidly metabolized in the intestinal mucosa and by first-pass hepatic metabolism to form the major metabolite of desogestrel is [etonogestrel] which is the biologically active metabolite. This modification is described by the hydroxylation in C3 of the desogestrel molecule. Later, etonogestrel is metabolized following the normal pathways of steroid metabolism. On the other hand, due to the 11-methylene side chain, desogestrel cannot be metabolized to other progestins. In addition to 3-keto-desogestrel, other phase I metabolites are 3alpha-OH-desogestrel, 3beta-OH-desogestrel, and 3alpha-OH-5alpha-H-desogestrel. These other metabolites are not known to have any pharmacologic effects, and are further converted in part by conjugation (phase II metabolism) into polar metabolites, mainly sulfates and glucuronides. Desogestrel is metabolized via hydroxylation and dehydrogenation to the active metabolite etonogestrel. Etonogestrel is metabolised via sulphate and glucuronide conjugation. Desogestrel is metabolized rapidly and completely in the liver and gut wall. It is metabolized to 3-keto-desogestrel, which mediates its progestogenic effects, and it is not metabolized further to another progestogen. The serum concentrations of 3-keto-desogestrel reached maximum levels within 2-3 hours after oral administration of desogestrel and were subsequently cleared with a half-life of 12-24 hours. The metabolism of desogestrel in microsomes from six hours livers in vitro /were studied/. The main metabolite formed was 3-keto-desogestrel; 3alpha-hydroxydesogestrel and 3beta-hydroxydesogestrel were also detected. The metabolism of desogestrel was inhibited by 50% by primaquine at a concentration of 30 umol/L, but not by levonorgestrel at 250 umol/L. For more Metabolism/Metabolites (Complete) data for DESOGESTREL (7 total), please visit the HSDB record page. Desogestrel has known human metabolites that include 3-beta-hydroxy-desogestrel, Desogestrel 17-O-glucuronide, and 3-alpha-hydroxydesogestrel. Biological Half-Life The terminal half-life of desogestrel is determined to be of 30 hours. Etonogestrel is eliminated with a mean half-life of approximately 30 hours, with no difference between single and multiple dosing. The elimination half-life for 3-keto-desogestrel is approximately 38+/-20 hours at steady state. /3-Keto-desogestrel/ - Absorption: Desogestrel is rapidly absorbed after oral administration in humans, with a time to reach maximum plasma concentration (Tmax) of 1-2 hours for the active metabolite 3-KD. The oral bioavailability of Desogestrel (based on 3-KD levels) is ~70-85% (due to first-pass metabolism in the liver, where Desogestrel is rapidly converted to 3-KD) [1][5] - Distribution: Desogestrel and 3-KD are highly bound to plasma proteins (>95%), primarily to sex hormone-binding globulin (SHBG) and albumin. In animal studies (rat, dog), 3-KD is widely distributed in tissues, with higher concentrations in the liver, uterus, and ovaries (tissue/plasma concentration ratio ~2-5 for uterus) [1][5] - Metabolism: The main metabolic pathway of Desogestrel is oxidation to 3-KD (catalyzed by CYP3A4 and CYP2C9 in humans), which is also biologically active (progestogenic activity similar to Desogestrel). 3-KD is further metabolized via hydroxylation and conjugation (glucuronidation, sulfation) to inactive metabolites [1][5] - Excretion: The elimination of Desogestrel metabolites (including 3-KD conjugates) is primarily via feces (60-70% of the dose) and urine (20-30%) in humans. The plasma elimination half-life (t1/2) of 3-KD is ~14-18 hours in humans, allowing for once-daily administration of Desogestrel-containing contraceptives [1][5] - Pharmacokinetic parameters in humans (oral Desogestrel 150 μg + EE 30 μg): Maximum plasma concentration (Cmax) of 3-KD: ~2.5-3.5 ng/mL; AUC0-24h of 3-KD: ~30-40 ng·h/mL; steady-state plasma concentrations are reached after 7-10 days of daily administration [5] |
| Toxicity/Toxicokinetics |
Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Desogestrel is only available in the United States in combination oral contraceptive products containing 150 mcg of desogestrel and 30 mcg of ethinyl estradiol. Based on the available evidence, expert opinion holds that nonhormonal methods are preferred during breastfeeding and progestin-only contraceptive are preferred over combined oral contraceptives in breastfeeding women, especially during the first 4 weeks postpartum. For further information, consult the record entitled, Contraceptives, Oral, Combined. ◉ Effects in Breastfed Infants A nonblinded, nonrandomized study compared oral desogestrel 75 mcg alone daily (n = 42) to an intrauterine device (IUD; n = 40) begun 28 to 56 days postpartum for contraception. No differences in infant length, weight or biparietal head circumferences were found after 1, 4, and 7 treatment cycles. Temporary breast enlargement was reported in 2 infants and increased sweating was reported in 1 infant in the desogestrel group, compared with no adverse effects reported in infants in the IUD group. The growth of some infants were again measured at 1.5 and 2.5 years; no clinically important differences were found. A breastfed (extent not stated) infant developed scrotal hair at 4 months of age. His mother had received the progestin, dydrogestrone, during the first trimester of pregnancy and began taking desogestrel 0.075 mg daily as a contraceptive beginning at 3 months postpartum. His mother discontinued desogestrel after 28 days and the scrotal hair resolved by 11 months of age. Desogestrel was a possible contributing cause of scrotal hair growth in this infant. ◉ Effects on Lactation and Breastmilk A nonblinded, nonrandomized study compared oral desogestrel 75 mcg alone daily (n = 42) to an intrauterine device (n = 40) begun 28 to 56 days postpartum for contraception. During the 7-month trial period, 1 woman dropped out of the trial because of diminished lactation compared with none in the IUD group. At the end of the first and fourth treatment cycle, there were no differences in the amount of milk produced between the desogestrel and IUD groups. No differences in triglyceride, protein or lactose content of milk were found at the end of 1, 4, and 7 cycles of therapy. A nonrandomized study followed 200 women given a desogestrel-only contraceptive 75 mcg daily for 6 months beginning at 6 weeks postpartum and compared them to 200 women who received placebos. No difference was found in the amounts of milk production or infant growth and development between the two groups. In a nonblinded, nonrandomized study in Türkiye of 4964 postpartum women were given the option of desogestrel 75 mcg (Cerazette) as a contraceptive starting at 21 days postpartum. On follow-up, the percentages of women who were breastfeeding at the third, sixth and ninth months postpartum were 68.4%, 54.8% and 58.5%, respectively. The authors concluded that the contraceptive had no negative impact on breastfeeding. Protein Binding The main metabolite of desogestrel is mainly found bound to albumin and sex-hormone binding globulin. Around 96-98% of the administered dose of desogestrel is found bound to plasma proteins from which 40-70% is found bound to sex-hormone binding globulin. - Lipid metabolism effects: Long-term administration (6 months) of Desogestrel (75 μg/day, progestogen-only pill) in healthy women did not cause significant changes in total cholesterol (mean change: -0.05 mmol/L vs. baseline), LDL-cholesterol (mean change: +0.03 mmol/L), or HDL-cholesterol (mean change: -0.02 mmol/L). A slight but non-significant increase in triglyceride levels was observed (mean change: +0.12 mmol/L), which was within the normal physiological range [4] - Bleeding side effects: In a study of women using Desogestrel-containing contraceptives (n=200), the incidence of spotting (light vaginal bleeding) was ~25% in the first month of treatment, decreasing to ~10% by the third month. Factors associated with increased spotting included younger age (<25 years) and previous use of non-hormonal contraceptives. No severe bleeding (requiring treatment discontinuation) was reported [3] - Thyroid function effects: In female Wistar rats treated with Desogestrel (0.1 mg/kg/day, 28 days), serum TSH levels increased slightly (mean: 1.8 mIU/L vs. 1.6 mIU/L in control), but FT3 and FT4 levels remained within the normal range (FT3: ~3.2 pmol/L vs. 3.3 pmol/L; FT4: ~15 pmol/L vs. 15.2 pmol/L). No histological damage to the thyroid gland was observed [7] - Plasma protein binding: Desogestrel and its metabolite 3-KD have high plasma protein binding (>95%). 3-KD binds to SHBG with higher affinity (binding rate ~80%) than to albumin (~15%), which may affect its distribution and biological activity in tissues with high SHBG expression (e.g., reproductive tissues) [5] |
| References |
[1]. In vitro and in vivo metabolism of desogestrel in several species. Drug Metab Dispos. 1998 Sep;26(9):927-36. [2]. Evaluation of hormonal suppression with two contraceptive regimens using ethinyl estradiol and desogestrel. Arch Gynecol Obstet. 2013 Feb;287(2):289-94. [3]. The effect of desogestrel, gestodene, and other factors on spotting and bleeding. Contraception. 1996 Feb;53(2):85-90. [4]. The effects of two progestogen-only pills containing either desogestrel (75 microg/day) or levonorgestrel (30 microg/day) on lipid metabolism. Contraception. 2001 Nov;64(5):295-9. [5]. Pharmacokinetic evaluation of desogestrel as a female contraceptive. Expert Opin Drug Metab Toxicol. 2014 Jan;10(1):1-10. [6]. Desogestrel down-regulates PHOX2B and its target genes in progesterone responsive neuroblastoma cells. Exp Cell Res. 2018 Sep 15;370(2):671-679. [7]. Effect of progestin on thyroid function in female Wistar rats. Front Endocrinol (Lausanne). 2024 Jun 5;15:1362774. |
| Additional Infomation |
Desogestrel is a 17beta-hydroxy steroid and a terminal acetylenic compound. It has a role as a contraceptive drug, a progestin and a synthetic oral contraceptive. Desogestrel, a prodrug, is a third generation progestogen and hence, a member of the gonane family which was largely used in Europe before being approved in the US and Canada. It was firstly generated from a study that showed that 11-beta and 11-alkylidene substituent in nortestosterone can enhance the biological activity. Desogestrel is now produced semi-synthetically from naturally occurred plant steroids. In the US, desogestrel is found only in combination with [ethinyl estradiol]. The first approved drug containing desogestrel was developed by Organon USA Inc in 1972 and FDA approved in 1992. Desogestrel is a Progestin. Desogestrel is a synthetic progestogen structurally related to levonorgestrel, with progesterone hormone receptor agonistic activity, used as a contraceptive and hormone replacement agent. Upon administration, desogestrel binds intracellular progesterone receptors in progesterone responsive tissue and the resultant complex interacts with DNA causing either gene transcription or gene repression. This eventually leads to an inhibition of gonadotropin releasing hormone (GnRH) secretion from the hypothalamus and a subsequent inhibition of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) release. This prevents ovulation and alters the cervical mucus. A synthetic progestational hormone used often as the progestogenic component of combined oral contraceptive agents (ORAL CONTRACEPTIVES, COMBINED). Drug Indication Oral desogestrel is used in combination with [ethinylestradiol] as a contraceptive agent for the prevention of pregnancy. Desogestrel is part of the combined oral contraceptives that contain a mix of estrogen and progestin which inhibit ovulation. FDA Label Mechanism of Action Desogestrel enters the cell passively and acts by binding selectively to the progesterone receptor and generating low androgenic activity. Its binding produces an effect like a transcription factor and thus, it produces modifications in the mRNA synthesis. The active metabolite of desogestrel, [etonogestrel], presents a combination of high progestational activity with minimal intrinsic androgenicity. Combination oral contraceptives act by suppression of gonadotropins. Although the primary mechanism of this action is inhibition of ovulation, other alterations include changes in the cervical mucus, which increase the difficulty of sperm entry into the uterus, and changes in the endometrium which reduce the likelihood of implantation. Receptor binding studies, as well as studies in animals, have shown that 3-keto-desogestrel, the biologically active metabolite of desogestrel, combines high progestational activity with minimal intrinsic androgenicity. The relevance of this latter finding in humans is unknown. In contrast to traditional progestogen-only pills, the contraceptive effect of Cerazette is achieved primarily by inhibition of ovulation. Other effects include increased viscosity of the cervical mucus. Recent studies have demonstrated that desogestrel activates the estrogen receptor-alpha at an activity of about 50% of that of 17beta-estradiol but activates the estrogen receptor-beta at an activity of only 20%. Desogestrel and/or its metabolite 3-keto-desogestrel (etonogestrel) were strongly progestogenic (approximately twofold over progesterone), weakly or not androgenic in animal studies in vivo and in-vitro binding assays and weakly or not active on the glucocorticoid receptor. The active metabolite of desogestrel, 3-ketodesogestrel, strongly bound to and activated progesterone receptor-A and, to a slightly lesser extent, progesterone receptor-B Improvement in oral contraceptive formulations was originally achieved through dose reduction of the estrogen and progestogen components. Recently, further improvement was achieved by increasing the selectivity of contraceptive progestins. The ratio between the affinity for the progesterone receptor and the affinity for the androgen receptor is an indicator of progesterone (or androgen) selectivity of a progestin. This ratio (selectivity index) reflects the relative amount of androgenic or progestational effect at a given dose. Relative selectivity can be characterized with in vitro receptor-binding studies and animal pharmacologic experiments. In comparison with levonorgestrel, desogestrel displays markedly lower androgenicity and slightly increased relative progestational activity. In receptor-binding experiments and animal pharmacologic studies, 3-keto-desogestrel, the active metabolite of desogestrel, shows the highest selectivity index. The favorable effect of desogestrel-containing oral contraceptives on lipoprotein metabolism and preexisting androgen-dependent skin disorders and the absence of adverse effects on blood pressure and body weight are attributed to the increased progestin selectivity of desogestrel. For more Mechanism of Action (Complete) data for DESOGESTREL (6 total), please visit the HSDB record page. - Contraceptive indication: Desogestrel is a synthetic progestin widely used in combined oral contraceptives (with EE) and progestogen-only pills (POPs) for female contraception. Combined regimens (e.g., EE 20-30 μg + Desogestrel 150 μg) inhibit ovulation by suppressing LH and FSH, thickening cervical mucus (to prevent sperm penetration), and altering the endometrium (to prevent implantation) [2][5] - Efficacy of contraceptive regimens: Two combined regimens (EE 20μg + Desogestrel 150μg and EE 30μg + Desogestrel 150μg) showed similar contraceptive efficacy (Pearl index <1 per 100 woman-years) in a 6-month study of healthy women (n=180). No significant difference in compliance (treatment discontinuation rate: ~5% in both groups) was observed [2] - Non-contraceptive potential: Desogestrel downregulates PHOX2B (a gene associated with neuroblastoma progression) in progesterone-responsive neuroblastoma cells, suggesting a potential role in the treatment of PR-positive neuroblastoma. However, this effect has not been validated in in vivo models or clinical trials [6] - Species differences in metabolism: The metabolic rate of Desogestrel is higher in rats (CLint: ~50 μL/min/mg protein) than in humans (CLint: ~20 μL/min/mg protein) and dogs (CLint: ~15 μL/min/mg protein). The major metabolite 3-KD is detected in all tested species, but minor metabolites (e.g., 6β-hydroxy-3-KD) vary by species [1] |
Solubility Data
| Solubility (In Vitro) |
DMSO: 16.7~62 mg/mL (53.7~199.7 mM) Ethanol: ~62 mg/mL (~199.7 mM) |
| Solubility (In Vivo) |
Solubility in Formulation 1: ≥ 1.67 mg/mL (5.38 mM) (saturation unknown) in 10% DMSO + 40% PEG300 + 5% Tween80 + 45% Saline (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 16.7 mg/mL clear DMSO stock solution to 400 μL PEG300 and mix evenly; then add 50 μL Tween-80 to the above solution and mix evenly; then add 450 μL normal saline to adjust the volume to 1 mL. Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH₂ O to obtain a clear solution. Solubility in Formulation 2: ≥ 1.67 mg/mL (5.38 mM) (saturation unknown) in 10% DMSO + 90% (20% SBE-β-CD in Saline) (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 16.7 mg/mL clear DMSO stock solution to 900 μL of 20% SBE-β-CD physiological saline solution and mix evenly. Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Solubility in Formulation 3: ≥ 1.67 mg/mL (5.38 mM) (saturation unknown) in 10% DMSO + 90% Corn Oil (add these co-solvents sequentially from left to right, and one by one), clear solution. For example, if 1 mL of working solution is to be prepared, you can add 100 μL of 16.7 mg/mL clear DMSO stock solution to 900 μL of corn oil and mix evenly. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 3.2209 mL | 16.1046 mL | 32.2092 mL | |
| 5 mM | 0.6442 mL | 3.2209 mL | 6.4418 mL | |
| 10 mM | 0.3221 mL | 1.6105 mL | 3.2209 mL |