Dalbavancin (MDL-63397; BI-397; A-A 1; VER-001; Dalvance; Xydalba), a semisynthetic lipoglycopeptide, is a novel, potent and second-generation lipoglycopeptide antibiotic active against gram-positive pathogens. It is in the same class as vancomycin which is the most widely used and one of the few treatments available to patients infected with methicillin-resistant Staphylococcus aureus (MRSA). Dalbavancin was designed based on vancomycin and teicoplanin. It possesses in vitro activity against a variety of Gram-positive pathogens including MRSA and methicillin-resistant Staphylococcus epidermidis (MRSE).
Physicochemical Properties
| Molecular Formula | C88H100N10O28CL2 |
| Molecular Weight | 1816.6918 |
| Exact Mass | 1814.608 |
| Elemental Analysis | C, 58.18; H, 5.55; Cl, 3.90; N, 7.71; O, 24.66 |
| CAS # | 171500-79-1 |
| Related CAS # | Dalbavancin hydrochloride;2227366-51-8;Dalbavancin-d6;1126461-54-8 |
| PubChem CID | 16134627 |
| Appearance | Typically exists as solid at room temperature |
| Density | 1.6±0.1 g/cm3 |
| Index of Refraction | 1.729 |
| LogP | 2.94 |
| Hydrogen Bond Donor Count | 21 |
| Hydrogen Bond Acceptor Count | 30 |
| Rotatable Bond Count | 22 |
| Heavy Atom Count | 128 |
| Complexity | 3740 |
| Defined Atom Stereocenter Count | 18 |
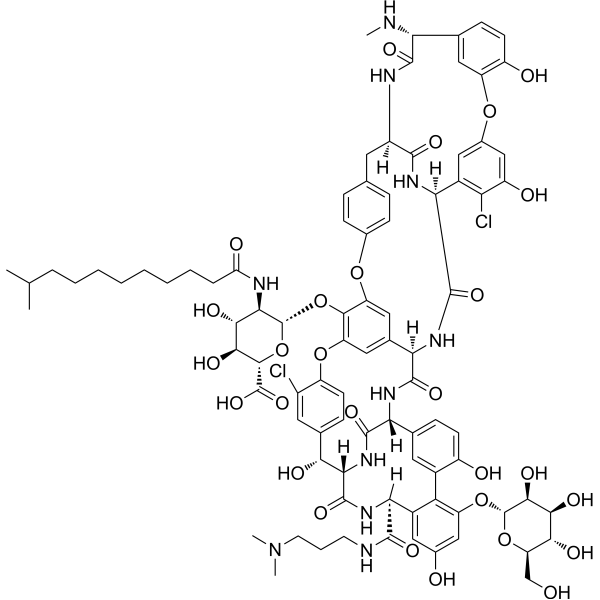
| SMILES | O[C@@H]([C@@H](O)[C@@H]1O)[C@H](O[C@@H]1CO)OC2=C(C3=CC([C@@](C4=O)([H])NC([C@@](NC([C@](NC([C@@](NC5=O)([H])C6)=O)([H])C(C=C7OC(C=C8[C@H]5NC)=C(C=C8)O)=C(C(O)=C7)Cl)=O)([H])C9=CC%10=C(O[C@H](O[C@H](C(O)=O)[C@@H](O)[C@@H]%11O)[C@@H]%11NC(CCCCCCCCC(C)C)=O)C(OC%12=CC=C6C=C%12)=C9)=O)=CC=C3O)C([C@@](NC([C@](N4)([H])[C@@H](C%13=CC=C(O%10)C(Cl)=C%13)O)=O)([H])C(NCCCN(C)C)=O)=CC(O)=C2 |
| InChi Key | KGPGQDLTDHGEGT-SZUNQUCBSA-N |
| InChi Code | InChI=1S/C88H100Cl2N10O28/c1-38(2)13-10-8-6-7-9-11-14-61(106)94-70-73(109)75(111)78(86(120)121)128-87(70)127-77-58-31-43-32-59(77)124-55-24-19-42(29-50(55)89)71(107)69-85(119)98-67(80(114)92-25-12-26-100(4)5)48-33-44(102)34-57(125-88-76(112)74(110)72(108)60(37-101)126-88)62(48)47-28-40(17-22-52(47)103)65(82(116)99-69)95-83(117)66(43)96-84(118)68-49-35-46(36-54(105)63(49)90)123-56-30-41(18-23-53(56)104)64(91-3)81(115)93-51(79(113)97-68)27-39-15-20-45(122-58)21-16-39/h15-24,28-36,38,51,60,64-76,78,87-88,91,101-105,107-112H,6-14,25-27,37H2,1-5H3,(H,92,114)(H,93,115)(H,94,106)(H,95,117)(H,96,118)(H,97,113)(H,98,119)(H,99,116)(H,120,121)/t51-,60-,64-,65-,66-,67+,68+,69+,70-,71-,72-,73-,74+,75+,76+,78+,87-,88+/m1/s1 |
| Chemical Name | Ristomycin A aglycone, 5,31-dichloro-38-de(methoxycarbonyl)-7-demethyl-19-deoxy-56-O-(2-deoxy-2-((10-methyl-1-oxoundecyl)amino)-beta-D-glucopyranuronosyl)-38-(((3-(dimethylamino)propyl)amino)carbonyl)-42-O-alpha-D-mannopyranosyl-N15-methyl- |
| Synonyms | MDL-63397; MDL 63397; Dalbavancin; dalbavancin B0; 171500-79-1; dalbavancine; Dalbavancina; A-A-1; VER-001; BI-397; VER001; MDL63397;A-A 1; BI397; BI 397; VER 001; MDL 63397; Dalbavancin. Dalbavancin B0; |
| HS Tariff Code | 2934.99.9001 |
| Storage |
Powder-20°C 3 years 4°C 2 years In solvent -80°C 6 months -20°C 1 month |
| Shipping Condition | Room temperature (This product is stable at ambient temperature for a few days during ordinary shipping and time spent in Customs) |
Biological Activity
| Targets | Glycopeptide; antibiotic |
| ln Vitro |
Dalbavancin is a semisynthetic lipoglycopeptide that is administered intravenously and was created to treat infections brought on by pathogens that are resistant to antibiotics. Strong in vitro bactericidal activity is demonstrated by dalapancin against gram-positive pathogens such as non-VanA strains of VRE, VISA, and S. aureus (MRSA). Dalbavancin has demonstrated greater potency against MRSA and β-hemolytic streptococci than other glycopeptide therapeutic agents, making it an excellent treatment for complicated skin and skin structure infections (cSSSIs)[1][2]. Dalbavancin susceptibility of B. anthracis strains. [1] Dalbavancin demonstrated potent in vitro activity. The MICs for the 30 strains of B. anthracis ranged from ≤0.03 to 0.5 μg/ml (Fig. 1), with the MIC50 being 0.06 μg/ml and the MIC90 being 0.25 μg/ml. In comparison, the vancomycin MICs ranged from 1 to 4 μg/ml. For the three MIC determinations, the MICs for the individual strains never varied by more than a single dilution. Antimicrobial susceptibility testing (AST) is performed to assess the in vitro activity of antimicrobial agents against various bacteria. The AST results, which are expressed as minimum inhibitory concentrations (MICs) are used in research for antimicrobial development and monitoring of resistance development and in the clinical setting for antimicrobial therapy guidance. Dalbavancin is a semi-synthetic lipoglycopeptide antimicrobial agent that was approved in May 2014 by the Food and Drug Administration (FDA) for the treatment of acute bacterial skin and skin structure infections caused by Gram-positive organisms. The advantage of dalbavancin over current anti-staphylococcal therapies is its long half-life, which allows for once-weekly dosing. Dalbavancin has activity against Staphylococcus aureus (including both methicillin-susceptible S. aureus [MSSA] and methicillin-resistant S. aureus [MRSA]), coagulase-negative staphylococci, Streptococcus pneumoniae, Streptococcus anginosus group, β-hemolytic streptococci and vancomycin susceptible enterococci. Similar to other recent lipoglycopeptide agents, optimization of CLSI and ISO broth susceptibility test methods includes the use of dimethyl sulfoxide (DMSO) as a solvent when preparing stock solutions and polysorbate 80 (P80) to alleviate adherence of the agent to plastic. Prior to the clinical studies and during the initial development of dalbavancin, susceptibility studies were not performed with the use of P-80 and MIC results tended to be 2-4 fold higher and similarly higher MIC results were obtained with the agar dilution susceptibility method. Dalbavancin was first included in CLSI broth microdilution methodology tables in 2005 and amended in 2006 to clarify use of DMSO and P-80. The broth microdilution (BMD) procedure shown here is specific to dalbavancin and is in accordance with the CLSI and ISO methods, with step-by-step detail and focus on the critical steps added for clarity [6]. |
| ln Vivo |
Treatment with Dalbavancin (15–240 mg/kg; intraperitoneal injection; every 36 or 72 hours; for 14 days; female BALB/c mice) results in an 80%–100% survival rate across all dose regimens[1].Bacillus anthracis, the causative agent of anthrax, can produce fatal disease when it is inhaled or ingested by humans. Dalbavancin, a novel, semisynthetic lipoglycopeptide, has potent activity, greater than that of vancomycin, against Gram-positive bacteria and a half-life in humans that supports once-weekly dosing. Dalbavancin demonstrated potent in vitro activity against B. anthracis (MIC range, < or =0.03 to 0.5 mg/liter; MIC(50) and MIC(90), 0.06 and 0.25 mg/liter, respectively), which led us to test its efficacy in a murine inhalation anthrax model. The peak concentrations of dalbavancin in mouse plasma after the administration of single intraperitoneal doses of 5 and 20 mg/kg of body weight were 15 and 71 mg/kg, respectively. At 20 mg/kg, the dalbavancin activity was detectable for 6 days after administration (terminal half-life, 53 h), indicating that long intervals between doses were feasible. The mice were challenged with 50 to 100 times the median lethal dose of the Ames strain of B. anthracis, an inoculum that kills untreated animals within 4 days. The efficacy of dalbavancin was 80 to 100%, as determined by the rate of survival at 42 days, when treatment was initiated 24 h postchallenge with regimens of 15 to 120 mg/kg every 36 h (q36h) or 30 to 240 mg/kg every 72 h (q72h). A regimen of ciprofloxacin known to protect 100% of animals was tested in parallel. Delayed dalbavancin treatment (beginning 36 or 48 h postchallenge) with 60 mg/kg q36h or 120 mg/kg q72h still provided 70 to 100% survival. The low MICs and long duration of efficacy in vivo suggest that dalbavancin may have potential as an alternative treatment or for the prophylaxis of B. anthracis infections.[1] Efficacy of Dalbavancin in the mouse inhalation anthrax model. (i) Postexposure prophylaxis model.[1] Dalbavancin, administered i.p. q36h (at ≥15 mg/kg) or q72h (at ≥30 mg/kg) for 14 days starting at 24 h postchallenge, provided significant protection (P < 0.001). Whereas all control mice died within 4 days postchallenge, 80 to 100% of the mice treated with dalbavancin survived with all dose regimens (Fig. 3). There was no indication of a dose-response relationship with the regimens of dalbavancin utilized. In comparison, as observed previously, 100% survival was obtained with a regimen of 30 mg/kg of ciprofloxacin twice daily for 14 days. (ii) Postexposure treatment model.[1] By 36 to 42 h postchallenge, clinical signs and deaths due to inhalational anthrax become evident in the mouse model. The efficacies of therapeutic agents, including ciprofloxacin and doxycycline, are significantly reduced when they are administered after that time. Delayed treatment with Dalbavancin provided 70 to 100% protection with i.p. administration of 60 mg/kg q36h or 120 mg/kg q72h starting at 36 or 48 h postchallenge (Fig. 4). In comparison, ciprofloxacin, administered at 30 mg/kg i.p. twice daily for 14 days beginning at 48 h postchallenge, protected 100% of the mice. Thus, intermittent dalbavancin treatment provided significant protection (P < 0.001 compared to the results for the controls), even when therapy was delayed until 36 or 48 h postchallenge, and its efficacy was at least comparable to that of ciprofloxacin administered twice daily (P > 0.05). The differences in survival obtained with ciprofloxacin treatment in this study and those obtained in previous studies are most likely due to variations in the final spore challenge dose within and between experiments that are inherent to the aerosol system. Due to this variation, the number of deaths prior to the initiation of the treatment at 48 h give different numbers of animals between groups at the initiation of treatment. The in vivo virulence of the S. aureus isolates was similar in the untreated control mice, based on the increase in thigh burden over the treatment period, i.e., 2.30 ± 0.14 log10 CFU/thigh. Two hours after infection, Dalbavancin was administered via the intraperitoneal route, with one of seven 2-fold-escalating doses of Dalbavancin (2.5, 5, 10, 20, 40, 80, and 160 mg/kg) being administered every 12 h for a 6-day treatment period. Untreated control groups were sampled at the start of therapy and at the end of the study. The thighs were removed from the animals and immediately processed for CFU determination. The results of these studies were analyzed by using a sigmoidal dose-effect model. The magnitude of the PK/PD index associated with each endpoint dose was calculated with the following equation: log10 D = log10 [E/(Emax − E)]/(N + log10 ED50), where E is the control growth for the static dose (D), E is the control growth − 1 log unit for D for 1-log kill, and E is the control growth − 2 log units for D for 2-log kill. Results of 1-log kill and 2-log kill were achieved against seven and six of the isolates, respectively (Fig. 2A and Table 2). The Dalbavancin in vivo exposure-response data were also considered relative to the PK/PD-linked driver AUC/MIC, using concentrations of free drug. Drug accumulation was calculated and included in AUC estimates. Using a sigmoidal Emax model, the data fit was strong for the seven-strain data set (R2 = 0.86), as shown in Fig. 2B. The numerical AUC/MIC values associated with each of the three treatment endpoints are also shown in Table 2. Net stasis was observed with a dalbavancin free-drug AUC (fAUC)/MIC value near 25. fAUC/MIC values near 50 and 100 were associated with 1-log and 2-log reductions, respectively, in organism burdens in the neutropenic mice. [5] |
| Enzyme Assay |
Susceptibility to Dalbavancin. [1] MICs were determined in triplicate by the broth microdilution method in cation-adjusted Mueller-Hinton broth (CAMHB), according to the methodology of the Clinical and Laboratory Standards Institute (CLSI) (11, 12). The final antibiotic concentrations were 0.03 to 64 μg/ml. After 18 to 24 h of incubation at 35°C, the MICs were determined both visually and spectrophotometrically (600 nm). The quality control strain Staphylococcus aureus ATCC 29213 was tested in parallel |
| Cell Assay |
Protocol [6] Note: Refer to CLSI documents M7-A10 and M100-S25 and/or ISO/FDIS 20776-1 for full details of the reference broth microdilution method for antimicrobial susceptibility. The reference methods allow for options in some of the procedures specific to inoculum preparation and MIC panel production to achieve the same end result. The method detailed here relates to one antimicrobial agent, Dalbavancin, and some of the steps represent one of potentially several ways the reference procedure can be done. Appropriate safety precautions (consistent with biosafety level 2) should be utilized10. The MIC panel format used for the purpose of this video publication is shown in Table 1. 1. Store Dalbavancin Powder [6] Upon receipt of diagnostic grade dalbavancin powder, store at -20 oC in a desiccated environment in a non-defrosting freezer. Prior to use, the powder should equilibrate to reach RT before opening. 2. Prepare MIC Panel Dilutions [6] Prepare a stock solution no higher than 1,600 µg/ml of dalbavancin in neat (pure) DMSO in sterile glass or plastic tubes, and use on the same day of preparation or store at -20 to -60 oC or below for future use in a non-defrosting freezer. Take into consideration the potency of dalbavancin as provided on the documentation received with the powder, when weighing the powder (see Equation 1 for example 800 µg/ml stock preparation). Dilute the stock dilution similar to the scheme as is shown in column 1 (“Source Concentration”) in Table 2 with neat DMSO in sterile glass or plastic tubes. Use one pipet for measuring diluent and another pipet for adding the initial dalbavancin stock to the first tube. For each subsequent dalbavancin stock concentration use a new pipet. Prepare 100X final MIC panel concentration dilutions (intermediate concentrations) with neat DMSO. As is shown in columns 2-4 of Table 2, combine appropriate volumes of source and DMSO to achieve desired intermediate concentration (volumes to be used will depend on number of MIC panels to be made). Prepare 0.004% P80: Prepare a fresh working stock solution of 2% P80 by adding 0.1 ml P80 to 4.9 ml dH2O. Sterilize by passing through a 0.22 micron filter and use solution on the same day of preparation. Prepare 0.004% P80 diluent by making a 1:500 dilution of 2% P80 (e.g., 0.3 ml of 2% P80 to 149.7 ml of CAMHB). Further dilute the intermediate concentrations prepared in Step 2.2 1:100 in cation adjusted Mueller Hinton broth (CAMHB) supplemented with 0.004% (v/v) polysorbate-80 (P-80) P-80 and/or LHB (for streptococci) added at double the final concentration because addition of inoculum (step 4.3) will result in a 1:2 dilution. See columns 6 and 7 in Table 2. 3. Prepare MIC Panels [6] Dispense 50 µl of each dalbavancin solution prepared in step 2.3 into appropriate wells of the MIC panel and include media only in one well (growth control well). A multi-channel pipet with sterile tips can be used for this step. Use panels immediately or seal with plastic film, place in plastic bags and immediately place in a non-defrosting freezer at ≤-20 oC (preferably at ≤-60 oC) until needed. If frozen panels are used, remove seals and place individual panels on lab bench for 15-30 min (until well contents are thawed) before proceeding to panel inoculation. 4. Inoculate MIC Panels, Perform Purity and Setup Colony Count [6] Select several well-isolated colonies from an 18-24 hr blood agar or other non-selective agar plate. Touch the top of each colony with a sterile loop or swab and transfer to 1-5 ml CAMHB or saline until turbidity is equivalent to a 0.5 McFarland standard. Assess turbidity by visual comparison to the 0.5 McFarland or with a photometric device. Within 15-30 min of preparation, dilute the inoculum 1:100 in CAMHB (100 µl into 10 ml CAMHB). For most bacteria tested against dalbavancin, with the exception of S. pneumoniae, this dilution will provide a final well concentration of 5 x 105 CFU/ml (acceptable range is 2-8 x 105 CFU/ml). For S. pneumoniae, bacterial concentration based on comparison of turbidity to a 0.5 McFarland is typically considerably less, therefore, dilute the inoculum 1:25 (400 µl into 10 ml CAMHB+10% +LHB). Within 15 min after inoculum preparation, transfer 50 µl of the final inoculum to each well (with exception of the sterility control well) of the MIC panel prepared in step 3. A multi-channel pipet using sterile tips can be used for this step. Perform a purity check by transferring and spreading a 1-10 µl aliquot from the positive growth control well using a sterile loop to a nonselective agar (e.g., trypticase soy agar with 5% sheep blood). Setup colony count by removing 10 µl from the growth control well with a single channel pipet and sterile tip and transfer to 10 ml of saline (1:1,000 dilution). Mix and transfer 100 µl with a single channel pipet and sterile tip to a suitable, nonselective agar medium (e.g., trypticase soy agar with 5% sheep blood) and spread over the entire agar surface with a sterile loop, repeating two times in different directions to assure even distribution of the inoculum (1:10 dilution). 5. Incubate MIC Panels, Colony Count and Purity Plates [6] Seal each MIC panel or stack of no more than 4 panels in a plastic bag, with plastic tape or with a tight-fitting plastic cover before incubating. Alternatively, place empty MIC panel on the top of stack of no more than 4 MIC panels, place a damp paper towel in a plastic container, place MIC panels in the plastic container and close container securely with lid. Incubate MIC panels in an ambient air incubator at 35 °C ± 2 °C for 16-20 hr (staphylococci and enterococci) and 20-24 hr (streptococci) within 30 min of inoculation. Incubate the colony count and purity plates under same conditions except incubate streptococci in a 5% CO2 incubator. 6. Read the MIC and Colony Count Plates; Check Purity Plate [6] Read the MIC as the lowest concentration that completely inhibits bacterial growth in the wells as detected by the unaided eye. Count colonies on the colony count plate. Multiply each colony by dilution factor (1:10,000) (e.g., 50 colonies is equivalent to 5 x 105 CFU/ml). An acceptable range is 20-80 colonies (2-8 x 105 CFU/ml) and is used as an approximate guideline. Check purity plate. If all colonies are similar to the colonies used in step 4.1, then the inoculum can be considered pure. If there are any other colonies present, then there is potential for a contaminant to be present in the MIC panel and the test should be repeated. |
| Animal Protocol |
Animal Model: Female BALB/c mice (6-8 weeks) challenged with Ames strain of B. anthracis[1] Dosage: 15 mg/kg, 30 mg/kg, 60 mg/kg, 120 mg/kg, 240 mg/kg Administration: Intraperitoneal injection; every 36 h or 72 h; for 14 days Result: Treatment started 24 hours after the challenge, with regimens of 15 to 120 mg/kg every 36 hours or 30 to 240 mg/kg every 72 hours. The rate of survival at 42 days showed an 80 to 100% efficacy. Pharmacokinetics.[1] Female ICR mice weighing 23 to 27 g were utilized. Single Dalbavancin doses of 5 and 20 mg/kg of body weight were administered i.p. or i.v. (via the tail vein) in 5% glucose solution. Blood samples were collected by cardiac puncture, while the mice were under halothane anesthesia, at 0.08, 0.25, 0.5, 1, 2, 4, 8, 12, 24, 48, 72, 96, and 144 h after dosing. Three animals per route, dose, and time point were utilized. The blood was collected in heparinized tubes, which were centrifuged to prepare the plasma. The plasma samples were stored at −20°C until analysis. Dalbavancin concentrations were determined by a microbiological agar diffusion assay that measures the total drug concentration, as described previously. The values of the PK parameters were determined by noncompartmental analysis. Efficacy studies.[1] Female BALB/c mice (age, 6 to 8 weeks) were challenged by aerosol with between 50 and 100 times the established 50% lethal dose (3.4 × 104 CFU) of a spore preparation of the Ames strain of B. anthracis. In the postexposure prophylaxis model, antibiotic treatment (administered in 0.2 ml i.p.) was initiated 24 h after challenge. Treatment groups (10 mice per group) received Dalbavancin once every 36 h (q36h) at doses ranging from 15 to 120 mg/kg or every 72 h (q72h) at doses ranging from 30 to 240 mg/kg for 14 days. A regimen of ciprofloxacin known to protect 100% of animals (30 mg/kg twice daily for 14 days) was tested in parallel. In the postexposure treatment experiments, the administration of Dalbavancin at 60 mg/kg q36h or 120 mg/kg q72h was initiated at later times (36 or 48 h) after challenge, when symptoms of infection could have appeared. Control mice received phosphate-buffered saline (PBS). The mice were monitored for survival for 42 days, at which time the surviving animals were killed and their organs were harvested to determine the tissue bacterial burden. For animals that died or that were moribund at earlier times, their organs were harvested at those times. Lungs, spleens, and the mediastinum region (lymph nodes) were aseptically removed, weighed, and homogenized in 1 ml of sterile water. Homogenates were serially diluted 10-fold in water, and 100-μl aliquots were plated on sheep blood agar. To determine the numbers of CFU of the anthrax spores, homogenates were heat shocked for 15 min at 65°C to kill vegetative cells, serially diluted, and plated as described above. The current studies were designed to define the pharmacodynamic (PD) target for Dalbavancin against S. aureus strains with dalbavancin MICs at or above the current FDA breakpoint (≥0.12 μg/ml), some of which were vancomycin-intermediate S. aureus (VISA) strains. The results from these studies provide a pharmacodynamic rationale in support of the current clinical dosing regimens. Furthermore, the data provide a starting point for the development of revised susceptibility breakpoints for this new compound. Seven strains of Staphylococcus aureus (including four vancomycin-intermediate S. aureus [VISA] strains) were studied (Table 1). The Dalbavancin and vancomycin MIC values were determined in triplicate using CLSI reference broth microdilution methods, in the presence of polysorbate 80. The Dalbavancin MIC range for the S. aureus isolates was 0.12 to 0.50 μg/ml. The neutropenic murine thigh infection model was used for all studies. Mice were inoculated with 107 CFU/ml of each strain. Single-dose plasma pharmacokinetic studies were performed with thigh-infected mice given intraperitoneal doses (0.2 ml/dose) of dalbavancin (2.5, 10, 40, 80, or 160 mg/kg). Dalbavancin plasma concentrations were measured with a liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay (Fig. 1); the lower limit of quantification for the assay was 0.05 μg/ml. Sample analysis precision (coefficient of variation [CV]) ranged from 5% to 6.4%, and accuracy (bias) ranged from −3.5% to −10.0%. Peak levels were observed by 2 to 6 h. Dalbavancin exhibited relatively linear pharmacokinetics, based on the dose-area under the concentration-time curve (AUC) relationship. The half-life was long and varied from 4.1 to 9.31 h. A protein binding value of 98.4%, based on prior studies in this model, was used. [5] |
| ADME/Pharmacokinetics |
Absorption, Distribution and Excretion In healthy subjects, dalbavancin AUC0-24h and Cmax both increased proportionally to dose following single intravenous (IV) dalbavancin doses ranging from 140 mg to 1500 mg, indicating linear pharmacokinetics. No apparent accumulation of dalbavancin was observed following multiple IV infusions administered once weekly for up to eight weeks, with 1000 mg on Day 1 followed by up to seven weekly 500 mg doses, in healthy adults with normal renal function. Following administration of a single 1000 mg dose in healthy subjects, an average of 33% of the administered dalbavancin dose was excreted in urine as unchanged dalbavancin and approximately 12% of the administered dose was excreted in urine as the metabolite hydroxy-dalbavancin through 42 days post-dose. Approximately 20% of the administered dose was excreted in feces through 70 days post-dose. Clearance and volume of distribution at steady state are comparable between healthy subjects and patients with infections. The volume of distribution at steady state was similar to the volume of extracellular fluid. 0.0513 L/h. Metabolism / Metabolites Dalbavancin is not a substrate, inhibitor, or inducer of CYP450 isoenzymes. Subsequently, metabolites have not been observed in significant amounts in human plasma. The metabolites hydroxy-dalbavancin and mannosyl aglycone have been detected in urine (< 25% of administered dose). The metabolic pathways responsible for producing these metabolites have not been identified; however, due to the relatively minor contribution of metabolism to the overall elimination of dalbavancin, drug-drug interactions via inhibition or induction of metabolism of dalbavancin are not anticipated. Hydroxy-dalbavancin and mannosyl aglycone show significantly less antibacterial activity compared to dalbavancin. Biological Half-Life Terminal half life is 346 hours. Dalbavancin PKs in mouse plasma.[1] The concentrations of dalbavancin (bound and unbound) in plasma at different sampling times after i.p. administration are shown in Fig. 2, and the values of the PK and the pharmacodynamic (PD) parameters for the 5- and 20-mg/kg doses are presented in Table 1. The peak plasma concentrations (Cmax) of dalbavancin, attained 2 h after i.p. administration, were 15.2 and 71.3 μg/ml with the 5-mg/kg and 20-mg/kg doses, respectively. The terminal half-life achieved with the 20-mg/kg dose was 53 h. At the dose of 20 mg/kg i.p., dalbavancin was detectable (≥0.4 μg/ml) in plasma for 6 days after administration. From 2 h on, the levels in plasma closely followed the kinetics obtained with i.v. administration of the same doses (Fig. 2). However, the areas under the concentration-time curves (AUCs; calculated to infinity) were somewhat lower after i.p. administration (176 and 848 mg·h/liter with the 5- and 20-mg/kg doses, respectively) than after i.v. administration (200 and 1,071 mg·h/liter, respectively; data not shown). As observed in other studies with animals and humans (6, 16, 27, 33, 34), the PKs of dalbavancin in mice were dose proportional, on the basis of comparisons of the AUCs achieved with doses of 5 and 20 mg/kg. After a single infusion of dalbavancin, the maximal plasma concentration (C max) and area under the plasma concentration-time curve (AUC) increased in a proportional manner from 500 mg to 1000 mg (C max: 157 μg/ml and 299 μg/ml; AUClast: 10,850 μg·h/ml and 22,679 μg·h/ml, on the 500-mg and 1000-mg regimens, respectively) with low inter-subject variability. The mean terminal phase half-life (t 1/2) was 204 and 193 h after the 500-mg and 1000-mg dose, respectively. Clearance and volume of distribution were similar for the two dose concentrations. Treatment-emergent adverse events reported were considered to be of mild intensity. There were no relevant changes in laboratory values or vital signs over time in subjects in either treatment group. Conclusions: Overall, dalbavancin 500 mg and dalbavancin 1000 mg, administered as a single 30-min infusion, was well tolerated in this population and resulted in plasma exposures similar to those in non-Asians.[3] |
| Toxicity/Toxicokinetics |
Effects During Pregnancy and Lactation ◉ Summary of Use during Lactation Dalbavancin is 93% plasma protein bound and is poorly absorbed orally, so it is not likely to reach the bloodstream of the infant or cause any adverse effects in breastfed infants. If dalbavancin is required by the mother, it is not a reason to discontinue breastfeeding. Monitor the infant for possible effects on the gastrointestinal tract, such as diarrhea, vomiting, and candidiasis (e.g., thrush, diaper rash). ◉ Effects in Breastfed Infants Relevant published information was not found as of the revision date. ◉ Effects on Lactation and Breastmilk Relevant published information was not found as of the revision date. Protein Binding Dalbavancin is reversibly bound to human plasma proteins, primarily to albumin. The plasma protein binding of dalbavancin is 93% and is not altered as a function of drug concentration, renal insufficiency, or hepatic insufficiency. |
| References |
[1]Antimicrob Agents Chemother. 2010 Mar;54(3):991-6.; [2]Ther Clin Risk Manag. 2008 Feb;4(1):31-40. [3]Clin Drug Investig. 2015 Dec;35(12):785-93. [4]J Antimicrob Chemother. 2016 Jan;71(1):276-8. [5]Antimicrob Agents Chemother. 2015 Dec;59(12):7833-6. [6]J Vis Exp. 2015 Sep 9:(103):53028. |
| Additional Infomation |
Pharmacodynamics The antibacterial activity of dalbavancin appears to best correlate with the ratio of area under the concentration-time curve to minimal inhibitory concentration (AUC/MIC) for Staphylococcus aureus based on animal models of infection. An exposure-response analysis of a single study in patients with complicated skin and skin structure infections supports the two-dose regimen for which dalbavancin injection is administered. Subsequently, the recommended dosage regimen of dalbavancin in patients with normal renal function is 1500 mg, administered either as a single dose, or 1000 mg followed one week later by 500 mg [FDA Label, F2356. Dalbavancin should be administered over 30 minutes by intravenous infusion. Furthermore, inn a randomized, positive- and placebo-controlled, thorough QT/QTc study, 200 healthy subjects received either dalbavancin 1000 mg intravenous (IV), dalbavancin 1500 mg IV, oral moxifloxacin 400 mg, or placebo. Neither dalbavancin 1000 mg nor dalbavancin 1500 mg had any clinically relevant adverse effect on cardiac repolarization. Dalbavancin is a semisynthetic glycopeptide used for the treatment of acute bacterial skin and skin structure infections caused or suspected to be caused by susceptible isolates of designated Gram-positive microorganisms including MRSA. It has a role as an antibacterial drug and an antimicrobial agent. It is a carbohydrate acid derivative, a monosaccharide derivative, a glycopeptide and a semisynthetic derivative. Dalbavancin is a second-generation lipoglycopeptide antibiotic that was designed to improve on the natural glycopeptides currently available, such as vancomycin and teicoplanin. Modifications from these older glycoprotein classes facilitated a similar mechanism of action for dalbavancin but with increased activity and once-weekly dosing. Its use is indicated for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by the following gram-positive microorganisms: Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), S. pyogenes, S. agalactiae, S. dysgalactiae, the S. anginosus group (including S. anginosus, S. intermedius, and S. constellatus), and Enterococcus faecalis (vancomycin susceptible strains). Dalbavancin acts by interfering with bacterial cell wall synthesis by binding to the D-alanyl-D-alanine terminus of nascent cell wall peptidoglycan and preventing cross-linking. Dalbavancin is a second-generation, semi-synthetic lipoglycopeptide antibiotic, with bactericidal activity against a variety of gram-positive bacteria. Upon administration, dalbavancin binds, at a site different from that of penicillins and cephalosporins, tightly to the D-alanyl-D-alanine portion of peptidoglycan chains, thereby preventing peptidoglycan elongation and interfering with bacterial cell wall synthesis. This leads to activation of bacterial autolysins and induces cell wall lysis. DALBAVANCIN is a Unknown drug with a maximum clinical trial phase of IV (across all indications) that was first approved in 2014 and has 3 approved and 7 investigational indications. Drug Indication Dalbavancin for injection is indicated for the treatment of adult patients with acute bacterial skin and skin structure infections (ABSSSI), caused by susceptible isolates of the following gram-positive microorganisms: Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus anginosus group (including Streptococcus anginosus, Streptococcus intermedius, Streptococcus constellatus) and Enterococcus faecalis (vancomycin susceptible strains). Dalbavancin is not active against gram-negative bacteria; therefore, combination therapy may be clinically indicated if the ABSSSI is polymicrobial and includes a suspected or documented gram-negative pathogen. To reduce the development of drug-resistant bacteria and maintain the effectiveness of dalbavancin and other antibacterial drugs, dalbavancin should be used only to treat infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy. Bacillus anthracis, the causative agent of anthrax, can produce fatal disease when it is inhaled or ingested by humans. Dalbavancin, a novel, semisynthetic lipoglycopeptide, has potent activity, greater than that of vancomycin, against Gram-positive bacteria and a half-life in humans that supports once-weekly dosing. Dalbavancin demonstrated potent in vitro activity against B. anthracis (MIC range, < or =0.03 to 0.5 mg/liter; MIC(50) and MIC(90), 0.06 and 0.25 mg/liter, respectively), which led us to test its efficacy in a murine inhalation anthrax model. The peak concentrations of dalbavancin in mouse plasma after the administration of single intraperitoneal doses of 5 and 20 mg/kg of body weight were 15 and 71 mg/kg, respectively. At 20 mg/kg, the dalbavancin activity was detectable for 6 days after administration (terminal half-life, 53 h), indicating that long intervals between doses were feasible. The mice were challenged with 50 to 100 times the median lethal dose of the Ames strain of B. anthracis, an inoculum that kills untreated animals within 4 days. The efficacy of dalbavancin was 80 to 100%, as determined by the rate of survival at 42 days, when treatment was initiated 24 h postchallenge with regimens of 15 to 120 mg/kg every 36 h (q36h) or 30 to 240 mg/kg every 72 h (q72h). A regimen of ciprofloxacin known to protect 100% of animals was tested in parallel. Delayed dalbavancin treatment (beginning 36 or 48 h postchallenge) with 60 mg/kg q36h or 120 mg/kg q72h still provided 70 to 100% survival. The low MICs and long duration of efficacy in vivo suggest that dalbavancin may have potential as an alternat[1] Increasing rates of antimicrobial resistance among strains of Streptococcus, Staphylococcus, and Enterococcus spp. have been widely documented. At least 50% of nosocomial Staphylococcus aureus infections in intensive care units in the US and UK are due methicillin-resistant S. aureus (MRSA). Drug resistance is not confined to hospitals, and community-acquired MRSA (CA-MRSA) strains are now common causes of complicated skin and soft-tissue infections (cSSTIs) in many regions. Dalbavancin is a novel parenterally administered semisynthetic lipoglycopeptide similar to the naturally produced glycopeptides vancomycin and teicoplanin. Dalbavancin features a multifaceted mechanism of action that inhibits bacterial cell wall formation by two different mechanisms that enhances its activity against a wide range of gram-positive bacteria including staphylococci, streptococci, enterococci, and some anaerobes. Additionally, dalbavancin possesses unique pharmacokinetic properties, the most significant of which is a long terminal half-life that allows for once weekly dosing. This attribute may prove to yield clinical and cost benefit. Overall, clinical trials indicate that dalbavancin is a safe, well-tolerated, and effective antimicrobial agent. In the largest investigation evaluating dalbavancin for the treatment of cSSTIs, it appeared to be as effective as linezolid. Dalbavancin, which is expected to receive FDA approval in 2008, appears to be a promising new antimicrobial agent for the treatment of cSSTIs. [2] Dalbavancin is a novel lipoglycopeptide with activity against Staphylococcus aureus, including glycopeptide-resistant isolates. The in vivo investigation reported here tested the effects of this antibiotic against seven S. aureus isolates with higher MICs, including several vancomycin-intermediate strains. Results of 1-log kill and 2-log kill were achieved against seven and six of the isolates, respectively. The mean free-drug area under the concentration-time curve (fAUC)/MIC values for net stasis, 1-log kill, and 2-log kill were 27.1, 53.3, and 111.1, respectively. [5] Animal models alone will of necessity provide the fundamental efficacy data needed for the development of therapeutics active against biological threat agents, such as B. anthracis. The mouse anthrax inhalation model is the first step in meeting the FDA's “animal rule” (46). The results obtained with dalbavancin in this model warrant further study with mice and strongly indicate a need to progress to studies with the nonhuman primate model of B. anthracis infection.[1] |
Solubility Data
| Solubility (In Vitro) | DMSO : ~100 mg/mL ( ~55.04 mM ) Water : ~100 mg/mL |
| Solubility (In Vivo) |
Note: Listed below are some common formulations that may be used to formulate products with low water solubility (e.g. < 1 mg/mL), you may test these formulations using a minute amount of products to avoid loss of samples. Injection Formulations (e.g. IP/IV/IM/SC) Injection Formulation 1: DMSO : Tween 80: Saline = 10 : 5 : 85 (i.e. 100 μL DMSO stock solution → 50 μL Tween 80 → 850 μL Saline) *Preparation of saline: Dissolve 0.9 g of sodium chloride in 100 mL ddH ₂ O to obtain a clear solution. Injection Formulation 2: DMSO : PEG300 :Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL DMSO → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Injection Formulation 3: DMSO : Corn oil = 10 : 90 (i.e. 100 μL DMSO → 900 μL Corn oil) Example: Take the Injection Formulation 3 (DMSO : Corn oil = 10 : 90) as an example, if 1 mL of 2.5 mg/mL working solution is to be prepared, you can take 100 μL 25 mg/mL DMSO stock solution and add to 900 μL corn oil, mix well to obtain a clear or suspension solution (2.5 mg/mL, ready for use in animals). Injection Formulation 4: DMSO : 20% SBE-β-CD in saline = 10 : 90 [i.e. 100 μL DMSO → 900 μL (20% SBE-β-CD in saline)] *Preparation of 20% SBE-β-CD in Saline (4°C,1 week): Dissolve 2 g SBE-β-CD in 10 mL saline to obtain a clear solution. Injection Formulation 5: 2-Hydroxypropyl-β-cyclodextrin : Saline = 50 : 50 (i.e. 500 μL 2-Hydroxypropyl-β-cyclodextrin → 500 μL Saline) Injection Formulation 6: DMSO : PEG300 : castor oil : Saline = 5 : 10 : 20 : 65 (i.e. 50 μL DMSO → 100 μLPEG300 → 200 μL castor oil → 650 μL Saline) Injection Formulation 7: Ethanol : Cremophor : Saline = 10: 10 : 80 (i.e. 100 μL Ethanol → 100 μL Cremophor → 800 μL Saline) Injection Formulation 8: Dissolve in Cremophor/Ethanol (50 : 50), then diluted by Saline Injection Formulation 9: EtOH : Corn oil = 10 : 90 (i.e. 100 μL EtOH → 900 μL Corn oil) Injection Formulation 10: EtOH : PEG300:Tween 80 : Saline = 10 : 40 : 5 : 45 (i.e. 100 μL EtOH → 400 μLPEG300 → 50 μL Tween 80 → 450 μL Saline) Oral Formulations Oral Formulation 1: Suspend in 0.5% CMC Na (carboxymethylcellulose sodium) Oral Formulation 2: Suspend in 0.5% Carboxymethyl cellulose Example: Take the Oral Formulation 1 (Suspend in 0.5% CMC Na) as an example, if 100 mL of 2.5 mg/mL working solution is to be prepared, you can first prepare 0.5% CMC Na solution by measuring 0.5 g CMC Na and dissolve it in 100 mL ddH2O to obtain a clear solution; then add 250 mg of the product to 100 mL 0.5% CMC Na solution, to make the suspension solution (2.5 mg/mL, ready for use in animals). Oral Formulation 3: Dissolved in PEG400 Oral Formulation 4: Suspend in 0.2% Carboxymethyl cellulose Oral Formulation 5: Dissolve in 0.25% Tween 80 and 0.5% Carboxymethyl cellulose Oral Formulation 6: Mixing with food powders Note: Please be aware that the above formulations are for reference only. InvivoChem strongly recommends customers to read literature methods/protocols carefully before determining which formulation you should use for in vivo studies, as different compounds have different solubility properties and have to be formulated differently. (Please use freshly prepared in vivo formulations for optimal results.) |
| Preparing Stock Solutions | 1 mg | 5 mg | 10 mg | |
| 1 mM | 0.5505 mL | 2.7523 mL | 5.5045 mL | |
| 5 mM | 0.1101 mL | 0.5505 mL | 1.1009 mL | |
| 10 mM | 0.0550 mL | 0.2752 mL | 0.5505 mL |